81 Treatment of Systemic Lupus Erythematosus
Clinical Course and General Treatment Strategy
The management of systemic lupus erythematosus (SLE) is challenging due to the clinical heterogeneity and the unpredictable course of the disease.1 SLE activity usually follows the flare pattern, which is characterized by a relapsing-remitting course. However, an equal number of patients have continuously active disease, and only a few have long periods of disease quiescence.2,3 Despite improvements in overall survival (85% to 90% during the first 10 years), some SLE patients are still at risk for premature death.4,5 Persistent inflammation inevitably results in irreversible major organ damage, which is linked to decreased quality of life and increased mortality.6 Accordingly, therapeutic strategies should aim at reducing overall burden of systemic inflammation. Achieving these goals requires (1) accurate assessment of disease activity and flares, (2) stratification according to severity of target organ involvement, (3) use of safe and effective drugs to induce remission promptly and prevent flares, and (4) prevention and management of disease and treatment-related comorbidities.7
In general, patients with mild lupus manifestations (skin, joint, and mucosal involvement) are treated with antimalarials or disease-modifying antirheumatic drugs (DMARDs), alone or in combination with low-dose oral glucocorticoids (GCs). Severe SLE with major organ involvement requires an initial period of intensive immunosuppressive therapy (induction therapy) to control aberrant immunologic activity and halt tissue injury, followed by a longer period of less intensive and less toxic maintenance therapy, to consolidate remission and prevent flares.8 Immunosuppressive therapy enables for the use of lower GC doses, thus reducing its deleterious effects.
SLE patients experience poor quality of life, which is only in part associated with disease activity and organ damage. Important contributors include fatigue, fibromyalgia, depression, and cognitive dysfunction.9,10 Treating physicians should regularly address these issues and engage symptomatic or remedial therapies as indicated. The realization that a significant proportion of patients features disease- and treatment-related comorbidities has shifted attention to adjunct therapies and primary prevention strategies such as renoprotective and cardiovascular disease risk reduction measures.
Patient and Physician Preferences
Health professionals are increasingly encouraged to involve patients in treatment decisions, recognizing them as experts with a unique knowledge of their own health and their preferences for treatments, health states, and outcomes.11 This approach may be particularly challenging in patients with severe lupus who may benefit from cytotoxic therapy, which is, however, associated with significant toxicity. This was demonstrated in a study of 93 well-educated women with mild lupus and good health status who were presented with descriptions of cyclophosphamide (CYC) and azathioprine (AZA) and were then asked to indicate their preferred choice of hypothetical treatment.12 The study patients had a strong preference for full disclosure of medication risks and treatment alternatives, and they preferred a collaborative role in decision making involving their health care. Nearly all (98%) participants chose AZA over CYC when both drugs conferred an equal probability of renal survival. Although most subjects switched preferences to CYC for better renal survival, a significant proportion (15% to 31%) were unwilling to switch to CYC for improved short- or long-term renal survival. Preference for the long-term benefits of CYC was greater among college than high school graduates, which could reflect a better understanding of the probabilities presented in the study among those with a higher education level. This underscores the importance of providing patients with tailored information to ensure that all patients accurately perceive the risks and benefits related to prescribed medications.
Drugs Used in the Treatment of Systemic Lupus Erythematosus
Key Points
There is an unprecedented array of promising biologic therapies currently in development in SLE.
Glucocorticoids
GCs exert broad inhibitory effects on immune responses mediated by T and B cells, as well as on the effector functions of monocytes and neutrophils. On the basis of these effects and their rapid onset of action, GCs have been remarkably efficacious in managing acute SLE manifestations. However, only two small randomized controlled trials (RCTs) have been conducted to demonstrate their efficacy in lupus.13,14
Tseng and colleagues15 examined the effect of a short course of moderate-dose GC in preventing flares in clinically stable but serologically active SLE. Severe flares, requiring increase in prednisone dose or addition of an immunosuppressive agent, or both, occurred in 6 out of 20 patients on placebo, as compared with none of the 21 patients who took prednisone (30 mg for 2 weeks, 20 mg for 1 week, and 10 mg for 1 week). The results agree with those of Bootsma and colleagues13 and suggest that in clinically stable but serologically active SLE patients, short-term GC therapy may avert a severe flare. This effect must be balanced against risks for overtreating patients with higher cumulative GC doses.
In severe, rapidly progressing disease or when doses greater than 0.6 mg/kg/day prednisone equivalent are required to control disease activity, GC pulse therapy may be introduced.16,17 Intravenous (IV) pulses of methylprednisolone (MP) (250 to 1000 mg daily for 1 to 3 consecutive days) are used, although there is no strong evidence to support a survival advantage. In addition to expediting remission, IV-MP pulses may allow for the use of lower GC doses during the induction period. In a trial from the National Institutes of Health (NIH), 82 patients with moderate to severe proliferative lupus nephritis (PLN) were randomized to receive sequential induction-maintenance therapy with IV-MP alone, IV-MP plus IV-CYC, or IV-CYC alone.18 Renal remission was significantly more common in both IV-CYC groups regardless of whether or not they received IV-MP. In the extended follow-up of the trial cohort, an analysis of protocol completers found that doubling of serum creatinine (SCr) was significantly lower in the combination group than in the IV-CYC group (relative risk [RR] 0.095).19 These findings indicate that combining IV-CYC with IV-MP in the treatment of lupus nephritis (LN) may confer an advantage in long-term renal outcomes without added toxicity.
Antimalarials and DMARD Therapy
Hydroxychloroquine
Antimalarial drugs, mainly chloroquine and hydroxychloroquine (HCQ), are commonly prescribed to SLE patients with skin and joint manifestations but are increasingly identified as an adjuvant treatment for achieving remission in severe lupus. A systematic review concluded that use of antimalarials resulted in a greater than 50% reduction in general SLE disease activity and to a moderate reduction in severe flares and GC dose.20 Beneficial effects on the lipid profile and subclinical atherosclerosis markers have also been described. Intriguingly, prospective observational studies have reported an inverse association between use of HCQ and accrual of irreversible organ damage, as measured using the Systemic Lupus International Collaborating Clinics–American College of Rheumatology Damage Index (SDI) (adjusted hazard ratio [HR], 0.73), and overall mortality rates (adjusted HR, 0.14 to 0.32).20 Although these findings may be confounded by milder forms of disease usually present in SLE patients who are prescribed antimalarials, the presumptive mode of action of antimalarials by inhibition of innate immunity pathways provides a plausible explanation for their multifold beneficial effects.21 HCQ is well tolerated with low rates of mild gastrointestinal and skin adverse events. Retinal toxicity is uncommon (estimated at 0.1% in patients who received HCQ for more than 10 years20), but routine ophthalmologic evaluation is recommended (annual screening during the first 5 years of usage is recommended for individuals who are treated with high HCQ dose [>6.5 mg/kg], for those treated more than 5 years, or for those who have other complicating factors).22
Methotrexate
Methotrexate (MTX), an antifolate agent commonly prescribed for rheumatoid arthritis (RA), has been used as steroid-sparing treatment for articular and cutaneous manifestations of SLE.23 MTX is administered weekly either orally or parenterally. Concomitant administration of folic acid (2.5 to 5 mg/week, not until 24 hours after the intake of MTX) is recommended to minimize toxicity (Table 81-1). Fortin and colleagues24 evaluated the efficacy of MTX in a 12-month placebo-controlled RCT in 86 SLE patients. Patients had mild to moderate disease, with musculoskeletal (93%), cardiovascular (74%), and hematologic (69%) manifestations. Approximately half of the patients in each group were on oral prednisone, whereas 41 patients in the placebo group versus 27 in the MTX group were on antimalarials. Among participants with comparable baseline prednisone dose, those on MTX received on average 1.33 mg/day less prednisone during the trial period compared with those in the placebo group. Fewer patients in the MTX group were also started on GCs (5% vs. 26% in the placebo group). MTX use was associated with a reduction in the mean during-trial Systemic Lupus Activity Measure (SLAM) score of 0.86 units. Together, these data suggest that MTX could be a reasonable alternative steroid-sparing agent in mild to moderate SLE.
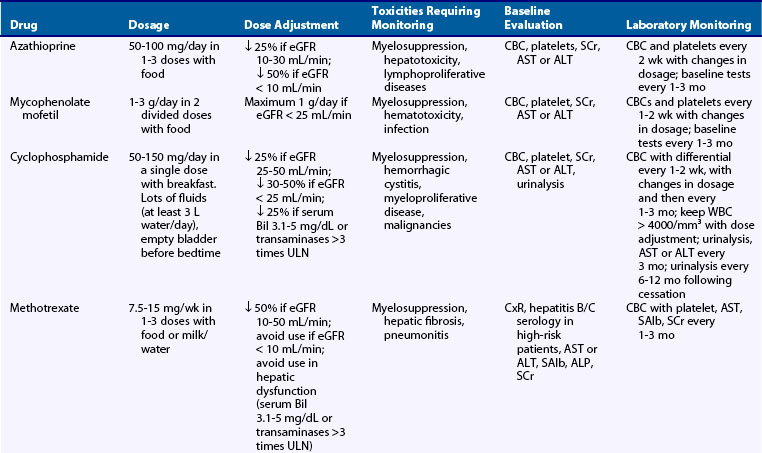
Leflunomide
Leflunomide, currently used in RA, has been used in LN refractory or intolerant to standard immunosuppressive therapy. Leflunomide requires a loading dose of 100 mg/day for 3 days followed by 20 mg/day thereafter. In a multicenter observational study, 110 patients with biopsy-proven PLN were assigned to either oral leflunomide or IV-CYC (monthly pulses 0.5 g/m2), both in combination with oral prednisone (0.8 mg/kg/day for 4 weeks, then tapered).25 After 6 months, complete and partial remission rates were 21% and 52% in the leflunomide group and 18% and 55% in the IV-CYC group, respectively. Repeat kidney biopsies showed significant reduction in activity index and pathologic transformation of 10 cases of diffuse to focal PLN. Similar rates of adverse events were observed in the two study groups and included mostly herpes zoster infection, alopecia, and hypertension. Better-designed RCTs are necessary to establish the efficacy, if any, of leflunomide in PLN. The drug is teratogenic and is contraindicated in patients who are trying to become or are pregnant.
Cytotoxic Therapy
Cyclophosphamide
Pharmacology and Route of Administration
The NIH trials demonstrated equivalent efficacy yet lower toxicity with monthly IV (0.5 to 1 g/m2) versus oral regimens, which led to the predominant use of IV-CYC in clinical practice (Table 81-4). Reversible myelotoxicity is common and dose related. After pulse therapy, the nadir of lymphocyte count occurs on days 7 to 10 and that of granulocyte count on days 10 to 14. The risk of infection increases with a white blood cell count less than 3000 cells/mm3, so the dose should be adjusted to keep it above this level (see Table 81-1). A prompt recovery from granulocytopenia usually occurs after 21 to 28 days. Thrombocytopenia is rare in CYC monotherapy. Reversible alopecia and nausea are common, whereas infections (especially herpes zoster), gonadal toxicity, and malignancy (including bladder toxicity and carcinoma) are less frequent though much more serious adverse events (see Comorbidities in Systemic Lupus Erythematosus and Women’s Health Issues later).
Table 81-4 National Institutes of Health Protocol for Administration and Monitoring of Pulse Intravenous Cyclophosphamide Therapy
CYC, cyclophosphamide; GFR, glomerular filtration rate; IV, intravenous; WBC, white blood cell count.
Use in Lupus Nephritis
RCTs with long-term follow-up have shown that intermittent pulse IV-CYC therapy is effective for moderate to severe PLN.33 CYC may retard progressive renal scarring, preserve renal function, and reduce the risk for the development of end-stage renal disease (ESRD) requiring dialysis or renal transplantation. Following induction therapy, a maintenance regimen is necessary to decrease the risk of flares.34 NIH studies have demonstrated that combination pulse therapy with IV-CYC and IV-MP improves renal outcomes without increasing toxicity.18,19 On the basis of these studies, the authors propose 7 monthly pulses of IV-CYC (0.5 to 1 g/m2) followed by quarterly pulses for at least 1 year beyond remission. For patients with moderate to severe disease, monthly pulses of IV-MP are given during the induction period.
Because of toxicity concerns and the appreciation that the disease may be less severe in whites, European investigators sought alternative IV-CYC protocols. In the Euro-Lupus Nephritis Trial involving mostly patients with milder forms of disease (mean SCr, 1.2 mg/dL; mean proteinuria, 3 g/day), less intensive regimens of IV-CYC (6 fortnightly pulses at a fixed dose of 500 mg each in combination with three daily doses of 750 mg of IV-MP) followed with AZA as maintenance had comparable efficacy and less toxicity than high-dose IV-CYC (8 pulses).35 Mean survival rate at 10 years was 92% for both groups; ESRD and doubling of SCr rates did not differ between the two groups.36 Therefore low-dose IV-CYC may be an alternative option for white patients with moderately severe LN.
Austin and colleagues37 compared cyclosporin A (5 mg/kg/day, then adjusted according to changes in SCr), IV-CYC (0.5 to 1 g/m2 × 6 monthly doses), and GC alone in an RCT in 42 patients with lupus membranous nephropathy (LMN) (median GFR, 83 mL/min/1.73 m2; median proteinuria, 5.4 g/day). All patients received alternate-day oral prednisone (1 mg/kg every other day for 8 weeks, then tapered to 0.25 mg/kg every other day). At 1 year, the cumulative probability of remission was 27% with prednisone, 60% with IV-CYC, and 83% with CsA. Rates of nephrotic syndrome relapse per 100 patient-months were 2.0 with CsA versus 0.2 with IV-CYC. Thus although IV-CYC and CsA are equally effective as induction therapy in LMN, CsA may require maintenance therapy (with lower doses of CsA, or AZA, or MMF) to prevent relapses.
Use in Extrarenal Disease
CYC has demonstrated efficacy in life-threatening extrarenal lupus manifestations such as severe thrombocytopenia (platelet count <20,000/mm3), neurologic disease, abdominal vasculitis, acute pneumonitis/alveolar hemorrhage, and extensive skin disease.38,39 Barile-Fabris and colleagues40 have reported superiority of IV-CYC against IV-MP for severe nonthrombotic neurologic SLE. In this trial, 32 patients were randomized to receive 3 pulses of 1 g of IV-MP followed by one of the following two treatments: pulses of 1 g IV-MP (monthly for 4 months, then every 2 to 3 months for 1 year) or IV-CYC (0.75 g/m2 monthly for 1 year and then every 3 months for another year). Seizures were the most common syndrome; other manifestations included peripheral neuropathy, optic neuritis, transverse myelitis, brain stem disease, coma, and internuclear ophthalmoplegia. Clinical response was observed in 18 of 19 patients who received IV-CYC as compared with 7 of 13 who received IV-MP. Thus the combination of pulses of IV-MP with IV-CYC is considered as treatment of choice for severe inflammatory neurologic SLE.
Other Agents
Chlorambucil
Chlorambucil (CAB) is an aromatic alkylating agent with substitution of the N-methyl group of mechlorethamine with phenylbutyric acid. The drug is given orally (0.1 to 0.2 mg/kg/day) with good absorption. The effects on immune functions are comparable with those described for CYC. Adverse events are also similar to those of CYC, except for bladder toxicity. More prolonged and less predictable bone marrow suppression can be observed. CAB has been associated with increased risk for leukemia. There is limited experience with the use of CAB in SLE, yet favorable outcomes in renal and extrarenal manifestations such as neuropsychiatric, vasculitis, and multiorgan involvement have been reported. In a retrospective study of 19 patients with predominantly LMN, Moroni and colleagues56 showed that CAB combined with alternate-month cycles of IV-MP was more effective than IV-MP alone in inducing remission of nephrotic syndrome (64% vs. 38%) and preserving renal function over a period of 83 months. However, the use of CAB for LMN or other lupus manifestations is limited.
Fludarabine
Fludarabine induces profound immunosuppression by depleting T and B lymphocytes and is used in the treatment of hematologic malignancies. A single pilot study in patients with active PLN was terminated prematurely due to severe hematologic toxicity.57 It is unlikely that fludarabine will be used in SLE.
Antimetabolites Calcineurin Inhibitors
Azathioprine
AZA interferes with the de novo synthesis of inosinic acid and inhibits the conversion of purine bases such as inosine to adenine and guanine ribonucleotides. AZA in doses of 2 to 2.5 mg/kg/day has been remarkably safe in the long term without significantly increasing the risk for infection, whereas it is associated with a marginally increased risk for malignancy. Gastrointestinal complaints are frequent, leading 15% to 30% of patients to discontinue the drug within 6 months. Mild liver enzyme elevation may occur, but severe liver injury is rare. Reversible, dose-related bone marrow toxicity is also common; leukopenia is encountered in approximately 4.5% and thrombocytopenia in 2% of patients receiving low-dose AZA (see Table 81-1). Notably, AZA toxicity is idiosyncratic and has been associated with genetic polymorphisms resulting in decreased thiopurine methyltransferase (TPMT) activity and impaired ability to detoxify intermediate metabolites.26 Concomitant use of allopurinol substantially increases AZA toxicity and should be avoided.
In lupus, manifestations such as mild PLN, thrombocytopenia with platelet count in the range of 20 to 50 × 103/mm3, and serositis may respond to AZA, usually in combination with moderate to high GC doses (Tables 81-2 and 81-3). Its efficacy as an induction-maintenance regimen has been tested in low-risk European patients with PLN who were randomized to receive pulse IV-CYC plus prednisone versus IV-MP plus AZA plus a tapering dose of prednisone.27 After 2 years, both groups received maintenance therapy with AZA plus prednisone. The two groups did not differ in terms of induction of remission, mean SCr and proteinuria levels during the first 2 years. However, after a median follow-up duration of 5.7 years, rates of doubling of baseline SCr and of renal relapses were higher in the AZA group. Thus the authors believe that AZA may be used in mild LN and in patients strongly opposed to CYC.
Table 81-2 Indications for Immunosuppressive Therapy in Systemic Lupus Erythematosus
| General Indications |
| Specific Organ Involvement |
| Renal |
| Hematologic |
| Pulmonary |
| Cardiac |
| Gastrointestinal |
| Nervous System |
Table 81-3 Recommended Immunosuppressive Therapy for Major Organ Involvement in Systemic Lupus Erythematosus
| Disease Severity | Induction Therapy | Maintenance Therapy |
|---|---|---|
| Mild | High-dose GC (0.5-1 mg/kg/day prednisone ×4-6 wk, tapered to 0.125 mg/kg every other day within 3 mo) alone or in combination with AZA (1-2 mg/kg/day) | Low-dose GC (prednisone ≤0.125 mg/kg on alternative days) alone or with AZA (1-2 mg/kg/day) |
| If no remission within 3 mo, treat as moderately severe | Consider further gradual tapering at the end of each year of remission | |
| Moderate | MMF (2 g/day) (or AZA) with GC as above; if no remission after the first 6-12 mo, treat as severe | MMF tapered to 1.5 g/day for 6-12 mo and then to 1 g/day; consider further tapering at the end of each year in remission Alternative: AZA (1-2 mg/kg/day) |
| Severe | Pulse IV-CYC alone or in combination with pulse IV-MP for the first 6 mo (background GC 0.5 mg/kg/day for 4 wk, then taper) | Quarterly pulses of IV-CYC for at least 1 year beyond remission |
| If no response, consider adding RTX or switch to MMF | Alternative: AZA (1-2 mg/kg/day), MMF (1-2 g/day) |
AZA, azathioprine; CYC, cyclophosphamide; GC, glucocorticoid; IV, intravenous; MMF, mycophenolate mofetil; MP, methylprednisolone; RTX, rituximab.
AZA has also been considered a safe and efficacious option for maintenance of remission in SLE including cases of moderately severe PLN.28,29 Two RCTs have compared AZA versus mycophenolate mofetil (MMF) as maintenance regimens in PLN. In the MAINTAIN trial, which included only European patients, both agents were equally efficacious in terms of time-to-renal flare, number of severe flares, renal remission, and doubling of SCr.30 In contrast, the ALMS trial, which included a larger number of patients with multiethnic backgrounds, reported increased renal flares in the AZA versus the MMF group (see Mycophenolate Mofetil later).31 To this end, both agents can be used as maintenance therapy on the basis of availability, clinical experience, and potential for pregnancy because MMF is associated with an increased risk of spontaneous abortion and fetal malformation. On the basis of their significant difference in cost, patients with mild to moderate LN, especially white individuals, could first be treated with AZA.32
Mycophenolate Mofetil
Pharmacology
MMF is a prodrug of mycophenolic acid, a potent inhibitor of inosine monophosphate dehydrogenase that is indispensable for the de novo synthesis of guanosine nucleotides. The lack of any salvage nucleotide synthesis pathway in lymphocytes renders them a selective target for MMF. Conversely, other tissues with high proliferative activity (skin, intestine, neutrophils) that have an intrinsic salvage guanosine synthesis pathway can escape the antiproliferative effects, which explains its more favorable toxicity profile compared with CYC. MMF has excellent oral bioavailability with peak levels occurring within 1 to 2 hours after administration and half-life of 17 hours. Although therapeutic drug monitoring to guide MMF dosing has been proposed in renal transplantation, validation of therapeutic MPA monitoring in LN is still required.41 Antacids and cholestyramine decrease the bioavailability of MMF, and the coadministration with AZA should be avoided. MMF may be teratogenic and should not be administered during pregnancy.
Use in Lupus Nephritis
Induction Therapy
Initial RCTs indicated equal or even superior efficacy of MMF over CYC in inducing remission in PLN, but their findings were limited by flaws in the design such as the low number of patients, the under-representation of severe forms of LN, and the short follow-up.28,42–44 The Aspreva Lupus Management Study (ALMS), one of the largest and most racially diverse RCT in LN that included a total of 370 patients, 27% of whom had estimated GFR less than 60 mL/min/1.73 m2, failed to demonstrate superiority of MMF (2 to 3 g/day) over monthly pulses of IV-CYC (0.5 to 1 g/m2).45 Both groups received oral prednisone, with a defined taper from a maximum starting dose of 60 mg/day. At 6 months, response rates were similar for both groups (56% for MMF, 53% for IV-CYC) and there were no differences in adverse events. Subsequently, three meta-analyses of RCTs have concluded that MMF is as effective as CYC (pooled relative risk [RR] for complete remission ranging 1.49 to 1.61) and has a better safety profile (pooled RR, 0.15 to 0.17 for amenorrhea; 0.41 to 0.78 for leukopenia; 0.77 to 0.83 for infections) as induction therapy for PLN.46–48 MMF may therefore be considered as induction therapy in moderately severe PLN, especially when gonadal toxicity is an issue.
MMF has demonstrated antiproteinuric effects in LMN,49 but there is lack of large RCTs to formally test its efficacy. A pooled analysis of 84 patients with pure LMN who participated in the ALMS trial45 and in the study by Ginzler and colleagues44 demonstrated comparable remission rates (percentage change of proteinuria and SCr were the primary end points) in MMF- and IV-CYC-treated patients.50 A trial testing MMF monotherapy in idiopathic membranous nephropathy failed to demonstrate efficacy.51 More data are necessary to delineate the role of MMF in the management of LMN.
Maintenance Therapy
Contreras and colleagues28 compared MMF versus AZA or quarterly pulses of IV-CYC as maintenance therapy in PLN following remission with 7 IV-CYC pulses. In this trial of 59 patients, 95% were black or Hispanic, 78% had diffuse PLN, with average SCr 1.6 mg/dL and serum albumin 2.7 mg/dL. After a follow-up of 72 months, MMF and AZA were superior to IV-CYC in terms of relapse-free survival (78% for MMF, 58% for AZA, 4% for IV-CYC), mortality, infections, and amenorrhea. This study was criticized for the insufficient number of patients to demonstrate superiority, the use of lower doses of IV-CYC, and the use of higher doses of GC, which may have decreased efficacy and increased the risk for infections. Two large multicenter RCTs have compared MMF versus AZA as maintenance regimen in PLN. The MAINTAIN Nephritis Trial included 105 European patients with moderately severe class III to IV PLN (10% had baseline SCr >1.4 mg/dL, 39% had proteinuria ≥3 g/day) who received induction therapy with 3 daily 750-mg IV-MP pulses followed by oral GC and 6 fortnightly IV-CYC pulses (500 mg per pulse).30 On the basis of randomization performed at baseline, AZA (target dose: 2 mg/kg/day) or MMF (target dose: 2 g/day) was given at week 12 to all patients. Over a 3-year period, the two groups did not differ in terms of time-to-renal flare, number of severe flares, renal remission, or doubling of SCr. Adverse events did not differ between the groups except for blood cytopenias, which were more frequent in the AZA group. These results are different from those reported for the ALMS maintenance part.31 This study included 227 patients (44% nonwhites) and showed a failure rate of 32% in the AZA group versus 16% in the MMF group at 3 years after successful induction therapy during the first part of the study with either MMF or IV-CYC. Differences in the study design and the induction protocol, the number and ethnicity of the included patients, and the outcome measures may account for the discrepant results. Of note, an RCT in ANCA-positive vasculitis found that MMF was less effective than AZA for maintaining disease remission following induction therapy with IV-CYC and GC.52 To this end, both MMF and AZA may be used for maintenance of remission in PLN on the basis of availability and potential for pregnancy.
Use in Extrarenal Lupus
A systematic review of open-label trials concluded that MMF may be effective for refractory skin and blood manifestations in SLE.53 A posthoc analysis of the ALMS trial data in LN patients showed that MMF was equally efficacious with IV-CYC pulse therapy (both in combination with tapered prednisone) on general disease activity.54 At week 24, BILAG-defined remission was achieved in the general (100% in MMF vs. 94% in IV-CYC), mucocutaneous (84% vs. 93%), musculoskeletal (91% vs. 96%), and hematologic (60% vs. 67%) domains. Normalization of C3/C4 and anti-dsDNA titers also occurred at a similar rate. Conversely, MMF showed no efficacy in preventing extrarenal flares in 75 patients who were followed at a single center for 5 years for renal (71%) or nonrenal disease.55 A substantial number of patients experienced flares during the second and third years of treatment, particularly in hematologic, mucocutaneous, and musculoskeletal domains. While awaiting additional data to define the efficacy of MMF in extrarenal lupus, the drug may be used in patients with moderately severe lupus who are intolerant or have not adequately responded to AZA.
Cyclosporin A
Pharmacology
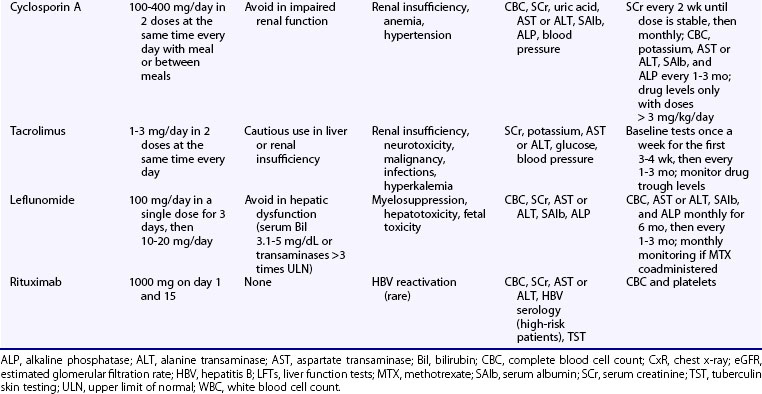
Cyclosporin A (CsA), a fungus-derived calcineurin inhibitor that deactivates T cells, also reduces antigen presentation and autoantibody production by lupus B cells. Following oral administration, peak serum concentration occurs within 1 to 8 hours. Drug concentration is measured in whole blood, but this is rarely necessary in autoimmune diseases, unless CsA is used in doses greater than or equal to 3 mg/kg/day. Clinical response occurs 1 to 2 months after treatment initiation. Several drugs interact with CsA, leading to reduced (rifampin, phenytoin, phenobarbital, nafcillin) or increased (erythromycin, clarithromycin, azoles, calcium channel blockers, amiodarone, allopurinol, colchicine) drug concentrations. The drugs may also augment CsA nephrotoxic effects (NSAIDs, aminoglycosides, quinolones, angiotensin-converting enzyme [ACE] inhibitors, amphotericin B). Common adverse events include mild gastrointestinal complaints, hirsuitism, gingival hyperplasia, and mild elevation in serum alkaline phosphatase levels. Tremor, paresthesias, electrolyte disturbances (hyperkalemia and hypomagnesemia), and hyperuricemia may also occur. Hypertension occurs in nearly 20% of patients receiving CsA and is controlled by either reduction of the dose or antihypertensive treatment. A major adverse effect is nephrotoxicity, which is reversible after adjustment of the dose or drug discontinuation, and CsA should be avoided in patients with impaired renal function (see Table 81-1).
Use in Proliferative Lupus Nephritis
Uncontrolled studies have demonstrated efficacy of CsA when used in combination with GC or in between quarterly doses of IV-CYC in refractory-to-conventional treatment PLN. Beneficial effects include reduction in proteinuria, stabilization of renal function, improvement in overall disease activity, and a modest steroid-sparring effect. Rihova and colleagues58 prospectively studied 31 LN patients (n = 24 with class III/IV nephritis) who were treated with CsA (5 mg/kg/day in two equal doses and then adjusted to trough level 80 to 120 ng/mL) and low-dose prednisone. After a mean of 7 months, all but two patients achieved complete response, defined as proteinuria less than 1 g/day and improved or stabilized renal function. About half of these patients, however, experienced a renal flare after CsA withdrawal.
Moroni and colleagues29 compared AZA with CsA as maintenance therapies in 69 patients with diffuse PLN and preserved renal function. All patients received induction therapy with 3 daily pulses of 1 g IV-MP, followed by prednisone and oral CYC for 3 months. They were then assigned to receive either CsA (4 mg/kg/day for 1 month and then tapered to 2.5 to 3 mg/kg/day) or AZA (1.5 to 2 mg/kg/day) for 2 to 4 years. The two groups did not differ in flare-ups, proteinuria, and blood pressure levels. Both agents were well tolerated. In another study, 40 patients with newly diagnosed PLN and mild renal insufficiency were randomly assigned to sequential induction and maintenance therapy with either CYC or CsA.59 The CYC regimen included 8 pulses IV-CYC (10 mg/kg) administered within 9 months, followed by 4 to 5 oral CYC boluses; CsA was given orally 4 to 5 mg/kg/day for 9 months and then tapered to 3.75 to 1.25 mg/kg/day within the next 9 months. Both groups received oral MP (0.8 mg/kg/day, then tapered). In the intention-to-treat analysis, 16 patients in the CYC group (76%) and 13 patients in the CsA group (68%) achieved complete or partial response at 9 months (induction phase). At the end of maintenance phase (18 months), the respective percentages were 52% for CYC and 95% for CsA. The trend for more favorable response in the CsA group was due to a higher proportion of patients achieving a 50% or greater decrease in proteinuria (38% in CYC group vs. 74% in CsA group). Despite its methodology flaws, this trial provides evidence for efficacy of CsA in mild to moderate proteinuric PLN with preserved renal function.
Use in Membranous Lupus Nephropathy
Balow and Austin37 performed an RCT in 42 patients with MLN to compare prednisone alone or in combination with CsA or monthly pulses of IV-CYC. Remission rates at 1 year were 27% for prednisone, 60% for IV-CYC, and 83% for CsA. During follow-up, however, rates of nephrotic syndrome relapse were 10-fold higher in the CsA than the IV-CYC group, suggesting that despite its effectiveness as induction therapy, CsA may require maintenance therapy to prevent relapses in MLN.
Use in Extrarenal Lupus
Uncontrolled studies of short duration have shown improvement in disease activity, anti-dsDNA titers, and cytopenias with modest GC reduction in SLE patients who received low-dose CsA.60,61 The BILAG group performed a multicenter, nonblinded RCT to compare the steroid-sparing effect of CsA (titrated to 2.5 to 3.5 mg/kg/day in two divided doses and adjusted to changes in SCr) versus AZA (2 to 2.5 mg/kg/day) in severe lupus requiring 15 or greater mg/day prednisone.62 Eighty-nine patients (66% whites) were randomized; 66% had active disease (defined as a BILAG A or B in any systems), and 34% entered the study because a different steroid-sparing agent was required. At 12 months, the unadjusted mean reduction in prednisolone dose was 9.5 ± 8.1 mg in the CsA group and 10.2 ± 6.2 mg in the AZA group. These improvements, however, were deemed as suboptimal for both drugs because almost 50% of patients with active disease at baseline failed to respond. The two groups did not differ in any of the secondary end points, namely disease activity, response to treatment, flares, damage accrual, and quality-of-life measures. In terms of safety, 23 patients (49%) who received CsA developed hypertension and another 6 patients (13%) showed a rise in SCr, both successfully managed by CsA dose reduction or addition of antihypertensive treatment. Thus CsA may be used as an alternative steroid-sparing agent to AZA in SLE patients, but with monitoring of the renal function and blood pressure.
Tacrolimus
Tacrolimus is a 10 to 100 times more potent calcineurin inhibitor than CsA. Systemic administration has been associated with dose-dependent reversible nephrotoxicity and blood pressure elevation, albeit less often than CsA. Other reported adverse effects include cardiomyopathy in children, anxiety, seizures, delirium and tremor, diabetes, and hyperlipidemia. In a pilot study of 10 SLE patients with skin and musculoskeletal disease, tacrolimus administered at doses of 1 to 3 mg/day for 1 year resulted in a significant reduction in SLE Disease Activity Index (SLEDAI) score and the dose of GC.63 Tacrolimus has also demonstrated beneficial effects in refractory-to-conventional therapy PLN and LMN.64–66 Miyasaka and colleagues67 conducted a placebo-controlled, double-blind trial in 63 patients with active mild-to-moderate nephritis requiring 10 or greater mg/day prednisone. They were randomized to receive a 28-week course of either tacrolimus (3 mg/day) or placebo in combination with GC (≤10 mg/day). In intention-to-treat analysis, only tacrolimus-treated patients had significant improvement in the author-defined nephritis disease activity index; 4 out of 27 patients in the tacrolimus group as compared with 1 out of 33 in the placebo group achieved proteinuria less than 0.3 g/day. Higher rates of GI toxicity and hyperglycemia were observed in the tacrolimus group.
Tacrolimus has been used in combination with MMF in severe or resistant LN. Cortés-Hernandez and colleagues68 prospectively studied 70 patients with moderately severe PLN who received induction therapy with 3 daily pulses of IV-MP (1 g/dose) and oral MMF (2 g/day), in conjunction with oral prednisone (1 mg/kg/day for 4 weeks, then tapered). In case of renal flare or treatment failure despite an increase in MMF dose, oral tacrolimus (0.075 mg/kg/day, adjusted to trough level 5 to 10 ng/mL) was added. At the end of the 65-month follow-up, tacrolimus had been started in 17 patients (24%), with 12 of them achieving complete or partial response after an average of 24 months. Tacrolimus/MMF combination therapy (tacrolimus 4 mg/day, MMF 2 g/day) has also been compared against IV-CYC (6 to 9 pulses of 1 g/m2) in patients with mixed class IV + V LN.69 In this study, patients had mean estimated GFR of 98 mL/min, proteinuria 4.4 g/day, and most had previously been treated with MMF or CYC. Both groups received 3 daily pulses of IV-MP (0.5 g/day) and then switched to oral prednisone. After 6 months, 10 patients in the “multitarget” group versus 1 patient in the IV-CYC group achieved complete remission. Combination therapy was well tolerated, and no major effects were observed. This is in contrast, however, with the significant toxicity reported in other patients with LN who received the same combination therapy.70 Therefore additional studies with a larger number of patients and longer follow-up are necessary to establish the efficacy, safety, and specific indications of such a multidrug approach in LN.
Biologic Therapies
B Cell–Depleting Therapies
Rituximab
Rituximab (RTX) is a chimeric mouse-human monoclonal antibody targeting the cell membrane protein CD20, which is expressed in all developmental stages of B cells, except for the hematopoietic stem cell and the plasma cell. Its mechanism of action involves cytotoxicity through complement activation, antibody-dependent cell-mediated cytotoxicity, and induction of apoptosis. RTX has been used in more than 450 SLE patients with refractory-to-cytotoxic therapy SLE, mainly as add-on therapy to GCs or other immunosuppressive agents, or both.71,72 Clinical response was noted in more than 80%, with manifestations such as neuropsychiatric disease,73 PLN, and autoimmune cytopenias, showing higher response rates. Data from a French lupus registry including a total of 136 patients also indicated response rates of at least 70% for various manifestations, with autoimmune cytopenias exhibiting the most favorable outcomes (85% for hemolytic anemia, 92% for thrombocytopenia). A clinically significant decrease in SLEDAI by 3 or more units was observed in 71%.74 With regard to LN, a meta-analysis of observational studies found an overall complete and partial response rate with RTX therapy of 69%, with lower rates observed in LMN.75 Two European cohorts, however, have reported comparable response rates in patients with PLN and LMN who received RTX in combination with steroids and IV-CYC.76
Unexpectedly, two RCTs failed to demonstrate efficacy of RTX in SLE. The EXPLORER trial assessed the effects of RTX over 52 weeks in patients with active moderate to severe extrarenal SLE.77 A total of 257 patients were randomized to receive RTX or placebo (two biweekly infusions of 1000 mg at baseline and at 6 months), in combination with background immunosuppression (AZA, MMF, MTX) and prednisone at a dose of 0.5 to 1 mg/kg/day, not tapered until day 16 after the first RTX infusion. At week 52, no difference was observed between RTX and placebo in the primary (BILAG-defined major or partial clinical response) or any of the secondary end points. The LUNAR trial evaluated the efficacy of RTX versus placebo, both in combination with MMF (3 g/day), in 144 patients with active PLN.78 By week 52, there was no significant difference between the two groups in terms of complete or partial renal response. Both studies were criticized for suboptimal design, mainly due to background therapies with high doses of oral GC and immunosuppressive drugs, which could have masked RTX effects. Alternatively, RTX may be more efficacious in patients with aggressive disease refractory to conventional treatment, as were most patients included in open-label trials.
RTX is generally well tolerated with mild infusion reactions being the most common adverse event, usually prevented by premedication with antihistamines and GCs. Mild infections are common (up to 20%), but the overall risk for serious or opportunistic infections is not significantly increased.79 Following the report of a few cases of progressive multifocal leukoencephalopathy (PML) in RTX-treated patients with RA and SLE, the U.S. Food and Drug Administration (FDA) has issued an alert about possible RTX and PML links. More data are necessary to evaluate whether RTX indeed increases the inherent risk for PML in SLE. Together, although RTX cannot be considered first-line therapy for mild to moderate SLE, its use may be justified in severe refractory cases on the basis of mounting evidence from open-label trials. Accordingly, the American College of Rheumatology (ACR) and the EULAR–European Renal Association have included rituximab (anti-CD20 mAb) as a therapeutic option for selected cases of LN refractory to conventional immunosuppressive treatment.80a,80b
Ocrelizumab
The BELONG trial evaluated the efficacy and safety of ocrelizumab (humanized anti-CD20 monoclonal antibody) versus placebo, both in combination with either MMF (up to 3 g/day) or IV-CYC (Euro-Lupus protocol) in 378 patients with PLN.80 The study was prematurely terminated due to increased rates of serious and opportunistic infections in ocrelizumab-treated patients, both when combined with MMF and IV-CYC.
B Cell Inhibitors
Epratuzumab
Epratuzumab is a recombinant anti-CD22 monoclonal antibody that modulates lupus B cell function.81 CD22 is a membrane co-receptor for the B cell receptor mediating inhibitory signaling through attenuation of calcium efflux. In 14 SLE patients with moderately active disease, epratuzumab therapy resulted in reduction in BILAG scores by greater than or equal to 50% in more than 70% of patients at 6 weeks and in nearly 40% of patients at 18 weeks.82 Drug tolerability was acceptable, and mild infections occurred in a minority of participants. Positive results have been announced for a phase IIb placebo-controlled, dose-defining trial in moderate to severe SLE. Using a composite BILAG-based response index, patients who received epratuzumab 600 mg weekly or 1200 mg biweekly achieved almost twofold higher response rates than the placebo group at the end of the 12-week treatment cycle. A similar rate of adverse events was recorded in the two groups.83 Phase III trials in moderate and severe SLE are under way.
Belimumab
The B lymphocyte stimulator protein (BLyS, also known as B cell activation factor [BAFF]) and the proliferation-inducing ligand (APRIL) are growth factors important for B cell survival and maturation. BLyS binds to three different B cell receptors, namely the transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI), the B cell maturation antigen (BCMA), and the BAFF receptor (BAFF-R), whereas APRIL signals through TACI and BCMA.84
Belimumab is a fully human anti-BLyS monoclonal antibody. An initial phase II trial of belimumab in combination with GCs and/or immunosuppressive agents in 449 active SLE patients did not meet its primary end points.85 In a posthoc analysis, however, serologically active patients (ANA titer ≥1 : 80 or anti-dsDNA positive) had a significantly higher response by week 52 in terms of SLEDAI and physician’s global assessment. These two parameters were combined with BILAG to define a novel activity index, the SLE responder index (SRI), which revealed favorable response rates for belimumab.86
Two subsequent phase III placebo-controlled RCTs (BLISS-52, BLISS-76) evaluated two different doses of belimumab (1 mg/kg, 10 mg/kg; dosed IV on days 0, 14, 28, and then every 28 days) on top of standard therapy in more than 1500 serologically positive SLE patients, using the SRI as the primary end point. Belimumab treatment resulted in a modest albeit significant reduction in disease activity and needs for additional GC, coupled with an increase in time-to-first flare.87 In BLISS-52, 58% achieved the primary end point in the high-dose belimumab arm versus 44% in the placebo group; the respective figures in BLISS-76 were 43% for belimumab and 34% for placebo. There were no significant differences in infectious adverse events between the two drugs. These data opened the way for approval of this agent for the treatment of SLE.
Atacicept
Atacicept (TACI-Ig) is a recombinant fusion protein of the extracellular domain of TACI and the human IgG1.Fc domain. TACI mediates signals from both BLyS and APRIL, thus affecting memory B cells, plasma cells, and immunoglobulin production. This was illustrated in two phase Ib placebo-controlled trials in patients with mild to moderate SLE.88,89 Both studies found no difference in adverse events between the two study groups, with the exception of mild injection-site reactions in patients receiving atacicept. However, a phase II/III trial of atacicept in combination with MMF in LN was prematurely terminated due to increased infection rates.
Co-stimulation Blockade
CD40-Ligand Blockade
CD40-ligand (CD40L) is expressed on activated T cells and stimulates antigen-presenting cells including B cells through engagement with CD40. Anti-CD40L treatment resulted in improvement of renal disease and increased survival in NZB/W F1 lupus mice.90 Two fully humanized monoclonal anti-CD40L antibodies have been developed for therapeutic trials in humans: BG9588 and IDEC-1310. The latter showed no efficacy in mild to moderate SLE.91 The encouraging results by the use of BG9588 (improvement in serologic activity and decrease in hematuria in 28 patients with PLN) were overpowered by the occurrence of serious thromboembolic events.92
Abatacept
Abatacept (CTLA4-Ig) is a recombinant protein that comprises the extracellular domain of human CTLA-4 fused to the Fc portion of human IgG1 and antagonizes CD28-mediated T cells. It is approved for the treatment of moderate to severe RA and juvenile idiopathic arthritis. CTLA4-Ig delayed disease progression in lupus-prone mice, especially when combined with CYC.93 A placebo-controlled, phase IIb trial of abatacept in combination with GC and background immunosuppressive therapy in 175 SLE patients with skin, joint, cardiovascular, and respiratory manifestations failed to meet its primary end point.94 After 1 year, the proportion of patients with new BILAG A/B flare after steroid taper did not differ between the two groups. However, the trial design may have undermined potential efficacy of abatacept because patients were started on high doses of GC (30 mg/day) and tapering began on day 29 or 57. Posthoc analysis showed that patients with polyarthritis benefited more from abatacept treatment. Serious adverse events were significantly more frequent in the abatacept group (20% vs. 7% in placebo) including bronchitis, diverticulitis, and gastroenteritis. Two RCTs are currently under way to evaluate abatacept in combination with MMF or IV-CYC in LN.
Anticytokine Therapy
Tumor Necrosis Factor Inhibitors
Tumor necrosis factor (TNF) has divergent effects on the immune systemic in lupus. In an open-label study, seven SLE patients with moderately active disease received four doses of 300 mg infliximab on day 0 and at weeks 2, 6, and 10, in combination with AZA or MTX. Anti-dsDNA and other autoantibodies increased in most patients, peaking 4 to 10 weeks after the last infliximab infusion but returning to baseline levels thereafter.95 In the prospective follow-up of 13 patients, 6 out of 9 patients with LN had a long-term (up to 5 years) response after four infusions of infliximab in combination with AZA.96 All five patients with severe arthritis responded, but only for 2 months after the last infusion. Long-term therapy was associated with high rates of serious adverse events. In another small open-label study in resistant proliferative or membranous LN, treatment with infliximab in combination with oral prednisone and CsA resulted in transient reduction of proteinuria and stabilization of renal function.97 Together, it is unlikely that TNF inhibition will be used routinely in SLE treatment.
Interferon Inhibition
Type I interferon (IFN-α) has been implicated in lupus pathogenesis through breakdown of immune tolerance. Serum IFN levels are increased and correlate with disease activity, and gene expression studies have identified an “interferon signature” in a subset of SLE patients.98 A phase I trial evaluated the effects of a single dose of anti-IFN-α monoclonal antibody (MEDI-545) in SLE.99 It was noted to downregulate IFN-inducible genes and other signaling pathways such as GM-CSF, TNF, IL-10, IL-1β, and BAFF. Two phase II trials are ongoing to assess safety and efficacy of anti-IFN therapy in lupus.
Anti-IL-6 Therapy
In lupus-prone mice, blockade of IL-6 or its receptor reduced anti-dsDNA levels, ameliorated proteinuria, and increased survival.100 Tocilizumab, a humanized monoclonal antibody against the α-chain of the IL-6 receptor, has shown efficacy in RA. Illei and colleagues101 tested tocilizumab in 16 patients with moderately active SLE, in combination with low-dose GC. Patients received tocilizumab (2, 4, 8 mg/kg) biweekly for 12 weeks and were monitored for an additional 8 weeks. Results revealed a correlation between clinical and serologic efficacy, evidenced by reduction in SLAM and SLEDAI indices, acute-phase reactants, and anti-dsDNA levels. Dose-dependent neutropenia (median decrease in neutrophil count by 56% in the 8 mg/kg group) and frequent infections (upper respiratory and urinary tract) were observed. To resolve issues on efficacy and safety, a larger trial is necessary.
Anti-IL-10 Therapy
IL-10 is upregulated in SLE and correlates with disease activity, although its exact pathogenic role has not been elucidated. An open-label trial in six steroid-dependent SLE patients showed improvement in disease activity reported up to 6 months after the administration of an anti-IL-10 murine monoclonal antibody (BN10) for 21 days.102 However, all patients developed antibodies against BN10, and new trials are awaited with a human anti-IL-10 monoclonal antibody.
Other Therapies
Intravenous Immunoglobulin
Intravenous immunoglobulin (IVIG) exerts immunosuppressive effects by interaction with anti-idiotypic antibodies, interference with the complement and cytokines, cytolysis of target cells, induction of apoptosis through Fc receptors, and modulation of co-stimulatory molecules.103 In an RCT of 14 patients, IVIG (400 mg/kg for 5 consecutive days monthly for 18 months) was as effective as pulse IV-CYC (1 g/m2 every 2 months for 6 months, then every 3 months for 1 year) as maintenance therapy of PLN.104 In another study, high-dose IVIG (2 g/kg divided over 5 days) in 1 to 8 monthly courses was administered in 20 SLE patients with cytopenias, massive proteinuria, arthritis, fever, arthralgia, mood changes, and psychosis. IVIG therapy resulted in improvement in SLEDAI and dose of GC.105 Common adverse events include fever, myalgia, headache, and arthralgia; less common are aseptic meningitis, nephropathy in patients who receive sucrose-containing preparations, and thromboembolic complications in older patients with atherosclerotic risk factors. The drug is contraindicated in IgA deficiency.
Synthetic Tolerogens
Tolerogenic peptides aim at restoring immune tolerance in lupus. Abetimus sodium (LJP-394) contains four identical dsDNA strands covalently linked to a small molecule platform. It is thought to reduce anti-dsDNA antibodies by binding and clearance of soluble anti-dsDNA antibodies and B cell receptor cross-linking on dsDNA-specific B cells. Initial studies showed decrease of anti-dsDNA levels, prolongation of the time-to-renal flare, and reduced renal flares, especially in patients with high-affinity anti-dsDNA antibodies.106–108 However, a phase III placebo-controlled trial in 317 LN patients failed to meet its primary end point (prolongation of time to renal flare), although abetimus-treated patients had 21% fewer flares, reduced proteinuria, and improved SLEDAI scores.109 The drug was well tolerated, and a 900-mg dosage was introduced in another trial, with no additional benefit.110 The spliceosomal peptide P140 is another tolerogen that has demonstrated benefit in early SLE trials. In a dose-escalation study in 20 patients with moderate SLE, the drug significantly decreased anti-dsDNA levels and slightly reduced SLEDAI.111 The results of an ongoing phase IIb trial using SRI as efficacy index are awaited.
Dehydroepiandrosterone
Dehydroepiandrosterone (DHEA) is a naturally occurring inactive steroid in adrenal glands, testes, and ovaries. Seven RCTs including a total of 842 patients have been performed to assess efficacy and safety of DHEA in SLE. Their meta-analysis showed that the drug had modest clinical effect only in mild to moderate disease.112 DHEA treatment also resulted in improvement in health-related quality of life measurements.
Management of Specific Systemic Lupus Erythematosus Manifestations and Treatment Algorithms
Mucocutaneous and Joint Disease
No single therapeutic agent has been officially approved for cutaneous lupus erythematosus (CLE). Mild malar rash and other photosensitive rashes usually respond to prophylaxis from sun exposure, but the use of sunscreens with high sun protection factor cannot be overemphasized. Topical GCs reduce redness and scaling. In an RCT of 78 patients with discoid lupus erythematosus (DLE), high-potency topical fluocinonide 0.05% was more effective than low-potency hydrocortisone 1% (response rates 27% vs. 10%, respectively).114 Calcineurin inhibitors (tacrolimus, pimecrolimus) are a useful alternative, especially for the face, because they cause less atrophic and rosacea-like effects than topical steroids.
In refractory to topical therapy skin disease, systemic antimalarials may be used alone or in combination with oral GCs (up to 20 mg/day). HCQ was equally effective and had more favorable safety profile than the oral retinoid acitretin in an RCT of 58 DLE patients.115 Other systemic treatment remains largely empiric. MTX has shown efficacy in refractory subacute CLE and DLE.116 Alternative choices include retinoids, dapsone, MMF, and IV-CYC.39 Thalidomide and IVIG should be reserved for patients with severe recalcitrant CLE due to potential serious neurotoxicity and high cost, respectively (Figure 81-1).117,118
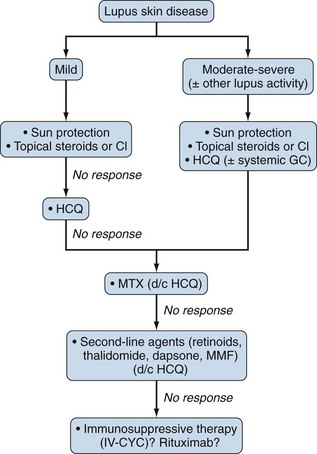
Lupus arthritis follows the pattern of nonerosive symmetric polyarthritis primarily affecting the small joints of the hands and feet. In mild arthritis, initial therapy should be based on antimalarials. In persistent or aggressive disease, utilization of DMARDs is advocated. In a prospective controlled study, SLEDAI scores, need for steroids, and articular involvement were significantly improved in SLE patients receiving MTX for 6 months.119 Leflunomide is used less often. Anti-TNF agents have been implicated for anti-dsDNA antibody development or even drug-induced lupus. RTX may be used in severe lupus arthritis refractory to DMARDs.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


