78 Enteropathic Arthritis
The gut wall is a leaky barrier exposed to commensal and pathogenic microorganisms.
Microbiota are essential for maturation and regulation of the immune system.
Genetic polymorphisms in Crohn’s disease can result in relative immune deficiency.
Some types of joint disease in inflammatory bowel disease are genetically determined.
HLA-B27 interacts with non–major histocompatibility complex genes.
Celiac disease is common in adults, 25% of whom have joint manifestations.
Patients with Whipple’s disease often present with joint symptoms.
Microscopic colitis is accompanied by extraenteric autoimmune manifestations.
This chapter deals with rheumatic conditions associated with gut pathology, which are commonly designated as enteropathic arthritis. People realized centuries ago that dysentery was sometimes followed by arthritis, a condition now known as reactive arthritis. Although it is a well-established example of enteropathic arthritis, reactive arthritis is covered in Chapter 76. This chapter covers musculoskeletal problems associated with inflammatory bowel disease (IBD), celiac disease (CeD), and other less common conditions. New insight into gut physiology, its barrier function, regulation of immune responses, and trophic functions is emerging. Knowledge regarding detailed interactions implicated in the generation of joint disease, however, remains incomplete. The fine-tuning of permeability, interactions between gut contents and gut mucosa, the nature of the gastrointestinal-associated lymphoid tissue, and how gastrointestinal-associated lymphoid tissue generates arthritis are at the core of this chapter.
Gut Biology and the Microbiota
Key Points
The gut mucosa is a leaky barrier and is covered by an “unstirred” layer.
Eighty percent of the gut wall consists of immune cells.
Gut permeability is delicately regulated and is disturbed in disease.
The gastrointestinal tract has three major biologic functions: serving as a barrier against hostile environmental factors; affecting fluid and food absorption and excretion of waste products; and producing major trophic host functions. The gut mucosa has an estimated surface area of 300 to 400 m2, which is 200 times the body’s skin surface area. This mucosa is constantly exposed to potentially harmful environmental agents against which defense is essential, but at the same time tolerance toward a normal microflora must be maintained. In order to fulfill these diverse functions, the gut is an immunologically privileged organ. Disturbances can lead to food allergy or inflammatory bowel disease (Figure 78-1).
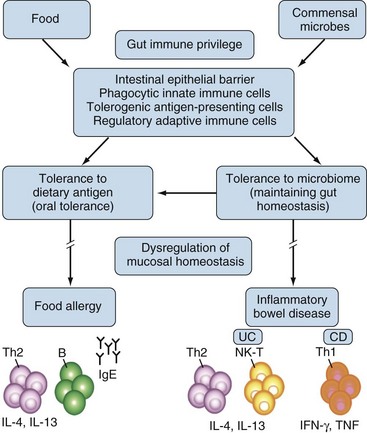
The gut epithelium constitutes a so-called leaky barrier. It consists of intestinal epithelial cells including Paneth cells in deep crypts, mucus-secreting goblet cells, M cells, and lymphocytes. It is covered by a thin layer of fluid separated from luminal flow and peristalsis of the intestine, the so-called unstirred layer, which slows down the diffusion of solutes and prevents loss of digestive enzymes. An increased thickness of the unstirred layer may contribute to malabsorption in CeD. Underneath the basal membrane is the lamina propria, which contains immune cells including lymphocytes, dendritic cells, and macrophages (Figure 78-2).1 The plasma membrane of the epithelial cells is impermeable to most hydrophilic solutes in the absence of specific transporters. The structure of the interepithelial paracellular space consists of the tight junction, the adherens junction, and the desmosome (see Figure 78-2B). The tight junction is considered rate limiting for permeability. A perijunctional actomyosin ring condensation is regulated by myosin light chain kinase (MLCK), which has been shown to have a central role in regulating tight junction transport and in tumor necrosis factor (TNF)-induced permeability increase.2 TNF inhibition will influence permeability via inhibition of MLCK transcription. Overexpression of MLCK, on the other hand, results in increased permeability and activation of immune cells in transgenic mice.3 Although initially healthy, these mice were prone to accelerated colitis if challenged.
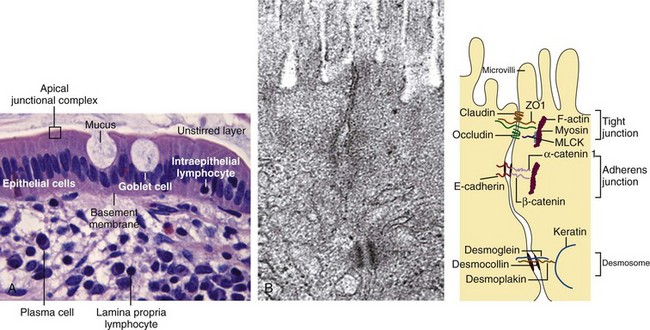
Na+ solutes smaller than 5000 daltons can pass through the epithelium freely, whereas bacterial products and dietary antigens are dependent on active transport mechanisms. Altered gut permeability can be observed in several diseases, among them nonalcoholic fatty liver disease.4 In this condition alterations in the zona occludens 1 (ZO1) expression result in perturbation of permeability. Genetic and exogenous factors such as drugs, nicotine, microorganisms,5 and cytokines are potential triggers of pathology by influencing paracellular functions in the gut epithelium.
Assessment of mucosal permeability and transport can be achieved by oral feeding of lactalbumin, lactoglobulin, polyethylene glycol particles, 51Cr-labeled ethylenediamine tetraacetic acid (EDTA), and sugars such as lactulose and mannitol, followed by urinalysis. In addition, intestinal permeability and function can be studied by regional perfusion with the help of endoscopic techniques that close off segments of the gut with inflatable balloons.6 A study applying enzyme-linked immunosorbent assays to fluid collected by this technique showed marked local immunity to a number of food-related antigens in patients with rheumatoid arthritis.7 Ethnic differences in gut permeability have also been described.8
The healthy gut harbors a mixture of native bacteria acquired at birth or shortly thereafter that retains a relatively constant composition; it also has a smaller population of transient bacteria of varying composition. The former are essential for health and live in symbiosis; the latter contain potential pathogens. Whereas the stomach and duodenum normally contain less than 103 mucosa-adhering bacteria, the number of bacteria increases to 104 in the jejunum and 107 in the ileum. Most of the latter group comprises gram-negative aerobic species. In the colon, the bacterial density is 1012 or more, consisting mostly of anaerobic bacteria. Transit time is fast in the upper gut and slow in the distal gut, but the immunologic impact of the microflora is higher in the proximal parts of the gut.9 In other words, the total number of bacterial cells called the human microbiota is 10 times that of cells in the body.10
Analysis of the 16S bacterial ribosomal RNA gene sequence has revealed the presence of a bewildering number of phylotypes in the human gastrointestinal tract, and the number of genes in the microbiome is estimated to be 100 times that of the human genome.11 There is also a high degree of diversity among healthy individuals and some correlation with obesity and other conditions. Most phylotypes have not been cultured, and their functions remain unknown. The potential pathogenicity of individual commensal microbes is demonstrated in a recent paper showing that a single species of segmental filamentous bacteria can trigger autoimmune arthritis in germ-free K/BxN mice by activating Th17 cells in lamina propria.12
The normal trophic functions of the gut require this microflora, as demonstrated by host defects in germ-free animals. Bacteria digest food carbohydrates into short-chain fatty acids, which facilitate the absorption of Ca2+, Mg2+, and Fe2+ ions; synthesize amino acids and vitamins; and secrete antibacterial protective substances. Some 300 to 500 different species are represented, and the composition is unique for each individual. Figure 78-3 shows some of the common colonic species and their functions.9
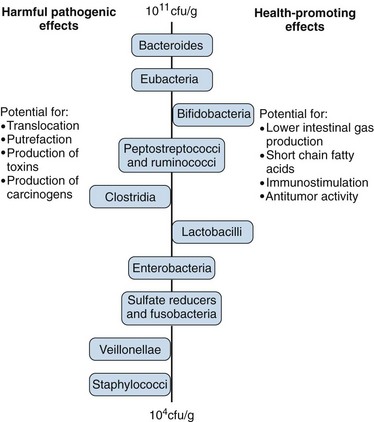
Figure 78-3 Physiologic roles of the intestinal microflora.
(From Salminen S, Bouley C, Boutron-Ruault MC, et al: Functional food science and gastrointestinal physiology and function, Br J Nutr 80(Suppl 1):S147–S171, 1998.)
Gastrointestinal-Associated Lymphoid Tissue and Its Interactions
Key Points
Microbiota are the major regulators of gastrointestinal-associated lymphoid tissue.
Retinoic acid promotes IgA and lymphocyte homing.
Flagellin stimulates dendritic cells to induce Th17 cells.
Vascular adhesion protein-1 is expressed in gut and synovium and is a putative therapeutic target.
Gut lymphocytes express α4β7, αEβ7, and CCR9, which are important for homing.
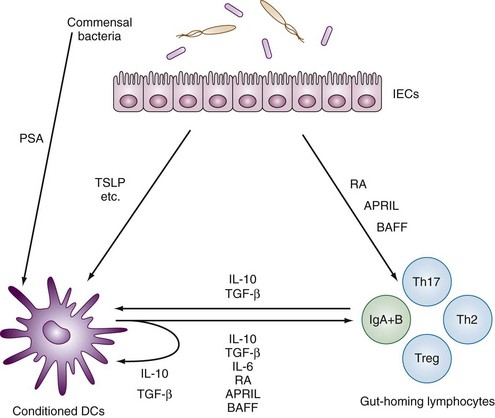
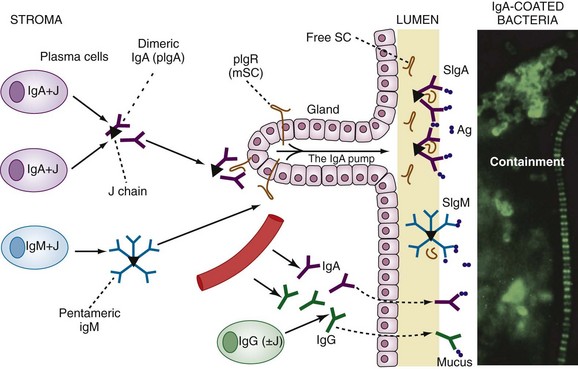
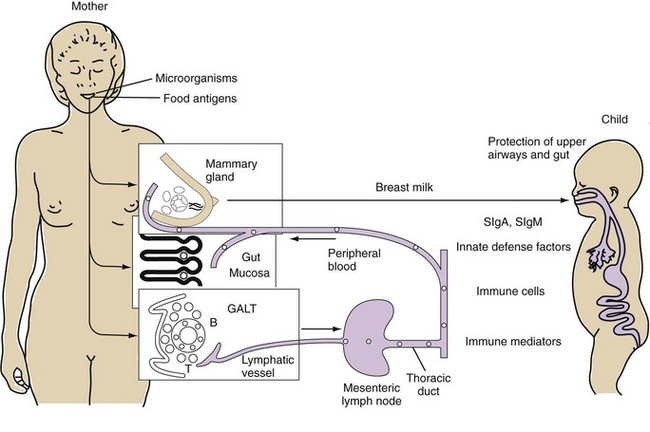
Gastrointestinal-associated lymphoid tissue (GALT) is a part of the mucosal immune system, a prominent feature of which is the dominating output of secretory immunoglobulin (Ig)A. This was discovered analyzing tears and saliva in the early 1960s.13 GALT is the largest lymphoid organ of the body, constituting 25% of the mucosal mass. Cellular components of GALT are localized in Peyer’s plaques, gut lymphoid follicles, lamina propria, and intraepithelial T cells. GALT is part of a complex defense and tolerance regulating system involving specialized dendritic cells (DCs) and intestinal epithelial cells (IECs) (Figure 78-4). Commensal bacteria interact with IECs, which send signals to DCs mediated by anti-inflammatory substances (e.g., thymic stromal lymphopoietin [TSLP]) and to lamina propria DCs. Bacteria also interact directly with DCs by means of their polysaccharide A. A subset of lamina propria DCs expresses CD11bhi, CD11chi, and Toll-like receptor (TLR5). When exposed to bacterial flagellin, these DCs promote the differentiation of T helper 17 (Th17) cells. These DCs also produce retinoic acid. IEPs also produce retinoic acid from dietary vitamin A, APRIL (a proliferation-inducing ligand), and BAFF. Retinoic acid stimulates the expression of the integrin α4β7 and CCR9, thereby influencing gut-homing lymphocytes.14 It was recently shown that Wnt–β-catenin signaling is required for the stimulation of expression of anti-inflammatory mediators by intestinal DCs.15 Bacteria can also penetrate into Peyer’s patches (PPs) and create a symbiotic environment with host cells.16 Cytokines such as interleukin (IL)-10 and transforming growth factor (TGF)-β serve anti-inflammatory functions. Retinoic acid, APRIL, and BAFF contribute to the dominating IgA production in local lymphoid cells. Gut enterochromaffin cells produce 5-hydroxytryptamine, which has anti-inflammatory effects and chromogranins with both proinflammatory and anti-inflammatory effects.17 Some of these not yet fully understood interactions are depicted in Figure 78-5. Plasma cells in the lamina propria become programmed to produce IgA, but the production is also skewed to formation of dimeric IgA, pIgA, in which the monomers are connected by joint, or J, chains.18 Intestinal epithelial glycoprotein pIgR, a polymeric immunoglobulin receptor that is also called secretory component (SC), is produced in IECs. It is a 100-kD transmembrane receptor for polymeric immunoglobulin, pIgA, and IgM. SC/pIgR is abundantly present in Peyer’s patches in the distal ileum. The pIgR in IECs combines with pIgA and to a lesser extent with IgM to form secretory IgA, SIgA, and SIgM, respectively, which are exported into the lumen and constitute a noninflammatory, non–complement-binding first line of defense. It is estimated that a healthy adult secretes 3 to 5 g of SIgA into the gut daily (Figures 78-6 and 78-7).18 Breastfeeding provides the newborn with abundant SIgA and SIgM, which confers passive protection and also regulates much of the child’s immune system19 (Figure 78-8).
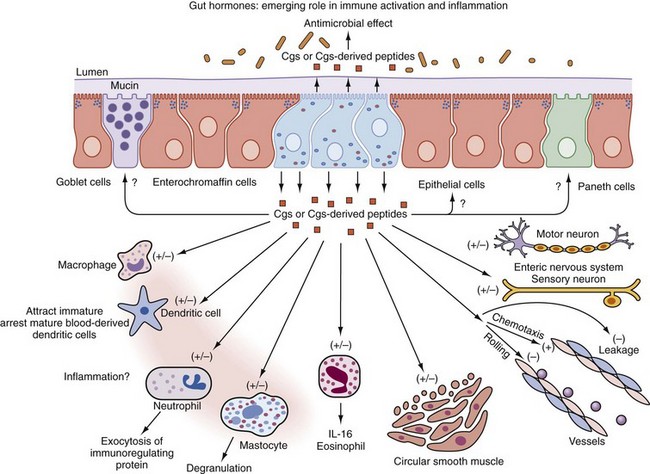
From the Peyer’s patches, primed B lymphocytes disseminate throughout the body’s mucous membranes, notably to other parts of the alimentary tract. Primed T lymphocytes also disseminate into the circulation and lymph nodes and home into target organs such as salivary glands (in Sjögren’s disease), lungs, and synovium.19 Vascular adhesion protein-1 (VAP-1) expressed on synovial epithelial cells is involved in lymphocyte homing, and P-selectin is a part of macrophage recruitment. VAP-1 is a bifunctional glycoprotein with both adhesive and amino-oxidative properties.20 Inhibition of this molecule is in development in oncology. A monoclonal antibody is protective against collagen-induced arthritis, and VAP-1 may become a target in the treatment of enteropathic arthritis. Most T lymphocytes in the mucosal lamina propria are CD4+, whereas intraepithelial T cells are mostly CD8+. Gut-associated lymphocytes preferentially express the integrins α4β7 and αEβ7 and the integrin receptor CCR9 on stimulation by intestinal dendritic cells.20
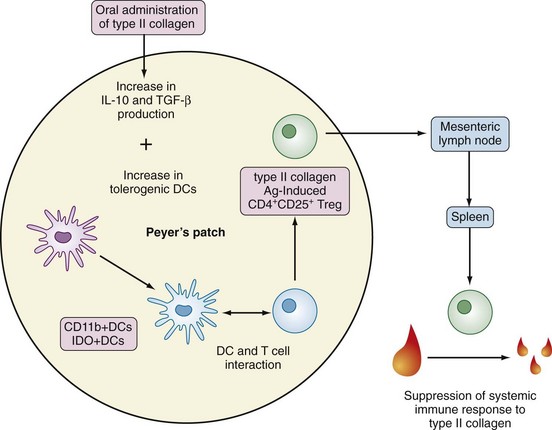
Induction of oral tolerance to type II collagen has demonstrated how GALT activation may ameliorate inflammatory joint disease (see Figure 78-7).21 A chain of events in the pathogenesis of enteropathic arthritis can begin with gastrointestinal infection with the appropriate microorganism in a genetically predisposed patient. This causes local inflammation in the gut mucosa, formation of secretory IgA, increased permeability, absorption of foreign material, and triggering of T lymphocytes. Circulating immune complexes and memory T cells localize to joints and cause synovitis (Figure 78-9). Figure 78-10 summarizes recent understanding of how colonization by segmented filamentous bacteria triggers arthritis in mice.
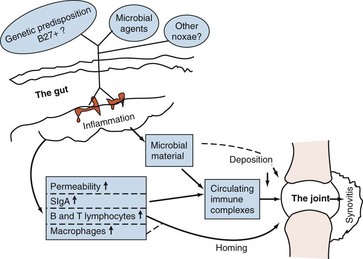
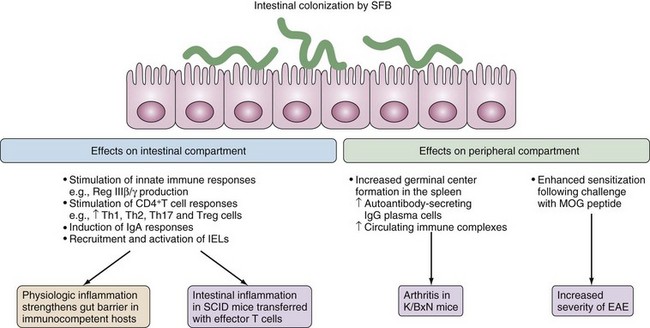
Figure 78-10 Effects of colonization of the gut immune system. Segmented filamentous bacteria (SFB) are spore-forming bacteria that are related to the genus Clostridium.51 Inherited from the mother microbiota, SFB develop strong interactions with the ileal mucosa and in immunocompetent mice, the bacteria can largely recapitulate the inducing effects of the whole microbiota on the postnatal maturation of the gut immune system. SFB induce the production of Reg IIIβ/γ microbicidal peptides,23,45 which protect against colonizing pathogens.46 Additionally, SFB simultaneously activate strong secretory IgA responses,44 induce the recruitment and activation of cytotoxic intraepithelial lymphocytes (IELs), and drive various T cell responses including a robust T helper 17 (Th17) cell response.28,45 In immunocompetent mice, SFB-induced proinflammatory and regulatory responses balance each other, which results in physiologic inflammation that strengthens the gut barrier. By contrast, colonization by SFB promotes the development of colitis in severe combined immunodeficient (SCID) mice that have been reconstituted with effector T cells.52 Intestinal colonization by SFB can also promote the development of inflammatory diseases outside of the gut. SFB promote arthritis in autoimmune nonobese diabetic (NOD) mice that express a transgenic T cell receptor (TCR) that is specific for a self-peptide (known as K/BxN mice), an effect ascribed to the induction of Th17 cells.54 SFB also enhance the severity of myelin oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalomyelitis (EAE). These aggravating effects may reflect the strong adjuvant properties of SFB.
(From Cerf-Bensussan N, Gaboriau-Routhiau V: The immune system and the gut microbiota: friends or foes? Nat Rev Immunol 10(10):735–744, 2010.)
Inflammatory Bowel Disease
Epidemiology
Key Points
The incidence and prevalence of inflammatory bowel disease are higher in developed societies.
Smoking increases the risk in both CD and ulcerative colitis.
Smokeless nicotine (snuff) may be inert or protective.
The prevalence and incidence of IBD are higher in developed countries. Demographics in developing countries, however, show an increase of IBD. Prevalence of Crohn’s disease (CD) is not decreasing, although immigration from minor developed countries is accelerating.22 The prevalence of CD and ulcerative colitis (UC) is about equal, and in the United States one observes between 50 and 100 cases per 100,000 population.5 In recent years, the incidence of UC has decreased in Western countries, whereas the previously low incidence of IBD in Eastern Europe, South America, and the Pacific has increased.22,23 This may be due in part to better reporting. Ethnic affiliation has an influence on the prevalence, although it is still unknown to what extent this is due to genetic or environmental factors. For example, the Jewish population has a higher susceptibility in general but prevalence approaches that of the background population. Romanies have a remarkably lower risk of developing IBD compared with the average Hungarian population.24,25 However, ethnic differences in the prevalence of extraintestinal manifestation have not been reported so far.
The incidence of CD and UC in a large Swedish population–based study was 11 per 100,000 and 18 per 100,000 person-years, respectively.26 In this study it was also shown that “ever smokers” had a relative risk of 1.5 (95% confidence interval [CI], 1.2 to 1.8) and 1.3 (1.1 to 1.5), respectively. It was also shown that “ever users” of moist snuff were not at increased risk of developing IBD. Therefore the nicotine component probably does not contribute to susceptibility. On the basis of the immunosuppressive signal mediated by nicotinic acetylcholine α receptors, smokeless nicotine may be innocent or indeed protective.27 The overall concordance in monozygotic twins is 36%, but it is only 16% in UC.28 The onset of disease is highest between 15 and 30 years of age, but IBD can start at any age. CD has a second peak later in life. There are no marked sex differences of IBD. Joint involvement has been reported in up to 25% of patients.6 Lower figures were reported from a large population-based Canadian study,29 but the study excluded peripheral arthritis, which may explain why less than 10% of patients had “arthritis.” Interestingly, asthma and multiple sclerosis were overrepresented in two studies of extraintestinal autoimmune manifestations of IBD (Table 78-1).30
Table 78-1 Extraintestinal Manifestations of Inflammatory Bowel Disease
| Feature or Disease | Crohn’s | Ulcerative Colitis |
|---|---|---|
| Peripheral arthritis | ≈15% | ≈10% |
| Axial or sacroiliac arthritis | ≈15%-20% | ≈10%-15% |
| Septic arthritis | Rare | Not reported |
| Skin | ||
| Erythema nodosum | Up to 15% | <15% |
| Erythema multiforme | Rare | ? |
| Pyoderma gangrenosum | 0.5%-2% | 0.3%-0.4% in severe disease |
| Aphthous ulcers | Rare | 1%-8% |
| Nephrolithiasis (oxalate) | <15% | ? |
| Amyloidosis | Very rare | Not reported |
| Liver disease | 3%-5% | 7% |
| Uveitis | 13% | 4% |
| Vasculitis | Takayasu’s | <5% |
| Clubbing of fingers | Yes | 1%-5% |
| Increased prevalence of asthma | Yes | Yes |
| Increased prevalence of multiple sclerosis | No | Yes |
Genetics
Key Points
At least 30 genetic associations have been identified in CD, and several occur in UC.
NOD2 (previously known as CARD15), IRGM, and ATG16L1 are associated with DCs.
IL23R and MDR1 are associated with both CD and UC.
IL23R deletion protects against colitis in mice.
CD has features of defective intracellular microbial handling.
Etiopathogenic research regarding IBD is in an active stage of development. New technology including genome-wide association studies (GWAS) has identified at least 31 genes showing replicated associations with CD31 and a similar number for UC.32 Approximately half of these associations are shared by both diseases. The strongest associations apart from human leukocyte antigen (HLA) are with the NOD2 (formerly known as CARD15) gene on chromosome 16 and the IL23R on chromosome 1. NOD2 and NOD1 are cytosolic sensors for the bacterial peptidoglycan muramyl dipeptide and trigger the synthesis of antibacterial α-defensins.33,34 Reduced mucosal expression of these defensins is found in patients with CD.35 IL23 signaling can result in activation of Th17 cells in the gut. The importance of IL23 is supported by the finding that IL23R −/− mice are resistant to induction of colitis.36 Also, IL23 is upregulated in patients with CD.37 Another association is found with the transcription factor STAT3, which is involved in Th17 activation. STAT3 is present on innate and reactive immune cells and is present in increased amounts in the gut mucosa.38 Another confirmed genetic association is that with ERAP1, an endoplasmic reticulum aminopeptidase previously known as ARTS1.39 ERAP1 functions by trimming the length of peptides before loading into the grove of HLA, and therefore it could play a role in the triggering of immune reactions in various locations such as gut and joint. A recent Spanish study looked at the Fc receptor gene FcRL-3, which is associated with susceptibility to RA, and found an association with peripheral arthritis in CD. Although this needs replication in other studies, it may indicate an example of a gene combination influencing disease phenotype.40 However, it is clear that no single gene or combination of genes alone can account for manifest IBD and clearly the intestinal microflora remains a major suspect as contributor to the cause of IBD, although final proof is lacking. Experimental models of IBD require presence of gut bacteria. Postoperative therapy with metronidazole has prolonged the time to relapse.14 Whereas CARD15 mutations were present in 43% of patients in the initial French study,41 later population studies found a much lower prevalence in northern European populations and no correlation in Asians. CARD15 mutations are not related to susceptibility in the United States. High expression of CARD15 messenger RNA has been found in the small intestine, and it is believed to be a regulator of nuclear factor κB (NFκB) signaling after the engagement of TLRs. IBD3 on chromosome 6 has shown the most constant association with IBD, and HLA-DRB1*0103 has been linked to severe UC in several studies.20 Further, a TNF microsatellite gene factor was associated with CD but not with UC. HLA-DR2 and DR3 associations have been linked to UC but not to CD.
Mutations of the detoxifying ATP-binding cassette, subfamily B, member 1 gene (ABCB1), also known as multidrug resistance 1 gene (MDR1), are strongly downregulated in unaffected colonic tissue of both CD and UC. TLR4 and TLR5 associations have been identified in several populations; this may be of special interest because they act in synergy with CARD15 and CARD4 in the induction of proinflammatory cytokines. TLR inhibitors are being investigated for therapeutic efficacy. Recently, researchers found a role for a virus as the contributing trigger in gene-manipulated mice, when mice with a disrupted autophagy gene, ATG16L1, were infected with a specific strain of murine norovirus and developed CD-like disease.37
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


