63 Anticytokine Therapies
Inhibition of a single key cytokine can be effective in autoimmune and inflammatory diseases.
Guarded optimism has been expressed regarding the long-term safety of TNF inhibitors.
Combining a TNF inhibitor with methotrexate achieves additive benefits.
IL-6 inhibition is an effective therapy in RA.
In recent years, discoveries delineating the immunopathophysiologic basis of various rheumatic diseases, combined with biopharmaceutical development, have allowed the introduction of biologic therapeutics. These agents target specific components of the immune response that are dysregulated and are thought to be central to the cause and sustenance of the disease process. In the rheumatoid synovium, for example, substantial evidence has been found of upregulation of key proinflammatory cytokines such as tumor necrosis factor (TNF), interleukin-1 (IL-1), IL-6, and others.1,2 Agents targeting these key mediators, in particular TNF, have considerable efficacy in the treatment of patients with rheumatoid arthritis (RA) and other systemic inflammatory disorders. The ability of TNF inhibitors not only to improve the signs and symptoms of disease but also to preserve functional status and quality of life and to inhibit disease progression has altered both physicians’ and patients’ expectations regarding antirheumatic treatment. Moreover, their success has driven research into the targeting of other cytokines relevant to the pathogenesis of autoimmune disorders. In this chapter, we focus on therapeutic agents that target TNF, IL-1, and IL-6.
Tumor Necrosis Factor Inhibitors
TNF plays a central role in the pathogenesis of RA and other inflammatory disorders. Although it can be produced by numerous cell types, in inflammatory conditions such as RA, TNF is produced largely by activated macrophages. Human TNF is synthesized and expressed as a 26-kD transmembrane protein on the plasma membrane and is cleaved by a specific metalloproteinase (TNF-converting enzyme). After proteolytic cleavage, TNF is converted to a 17-kD soluble protein, which oligomerizes to form the active homotrimer. The actions of TNF are mediated through two structurally distinct receptors: TNF-RI (55 kD; CD120a) and TNF-RII (75 kD; CD120b).3 The two receptors differ in their binding affinities signaling properties, and primary functions.3,4 The binding of TNF to its receptor can initiate several signaling pathways. Signaling cascades include the activation of transcription factors (e.g., nuclear factor κB [NFκB]), protein kinases (intracellular enzymes that mediate cellular responses to inflammatory stimuli, such as c-JunN-terminal kinase [JNK] and p38 mitogen-activated protein [MAP] kinase), and proteases (enzymes that cleave peptide bonds, such as caspases).
TNF may contribute to the pathogenesis of RA through myriad mechanisms, including induction of other proinflammatory cytokines (e.g., IL-1, IL-6) and chemokines (e.g., IL-8); enhancement of leukocyte migration by increasing endothelial layer permeability and adhesion molecule expression and function; activation of numerous cell types; and induction of the synthesis of acute phase reactants and other proteins, including tissue-degrading enzymes (matrix metalloproteinase enzymes) produced by synoviocytes or chondrocytes. The pivotal role of TNF in mediating such diverse inflammatory activities provided the rationale for targeting this cytokine in systemic inflammatory diseases.5 Initially, animal studies proved that inhibition of TNF with monoclonal antibodies or soluble TNF-R constructs ameliorated the signs of inflammation and prevented joint destruction.6 Subsequently, studies in humans confirmed the substantial efficacy of these compounds.
Although all five available agents are macromolecule TNF inhibitors, differences among them have been noted.7 The monoclonal antibodies infliximab, adalimumab, golimumab, and certolizumab pegol are specific for TNF, whereas etanercept binds both TNF and lymphotoxin-α (LT-α; previously referred to as lymphotoxin). Given intravenously, infliximab has a high peak concentration followed by steady-state elimination, whereas etanercept, adalimumab, golimumab, and certolizumab pegol, because they are given subcutaneously, have “flatter” pharmacokinetic profiles. With the exception of certolizumab pegol, these agents are capable of affecting Fc-mediated functions, such as complement-dependent cytolysis and antibody-dependent cell-mediated cytotoxicity, and all bind to both soluble and membrane forms of TNF, although some relative differences in affinity may be noted. Other differences, such as effects on cytokine secretion, have been observed in some in vitro studies.8 Regarding apoptosis, the data have been somewhat discrepant. In patients with RA, both the anti-TNF monoclonal antibody infliximab and the soluble receptor construct etanercept are capable of inducing apoptosis in synovial macrophages.9 However, in patients with Crohn’s disease, etanercept was not clinically effective at the doses studied and did not induce apoptosis. In contrast, the anti-TNF monoclonal antibodies infliximab and adalimumab were clinically effective and induced apoptosis in highly activated lymphocytes.10 However, certolizumab pegol is effective in Crohn’s disease and is not able to induce apoptosis. The extent to which these potential differences among TNF inhibitors correlate with any specific aspects of efficacy or toxicity remains to be established.
Infliximab
Structure
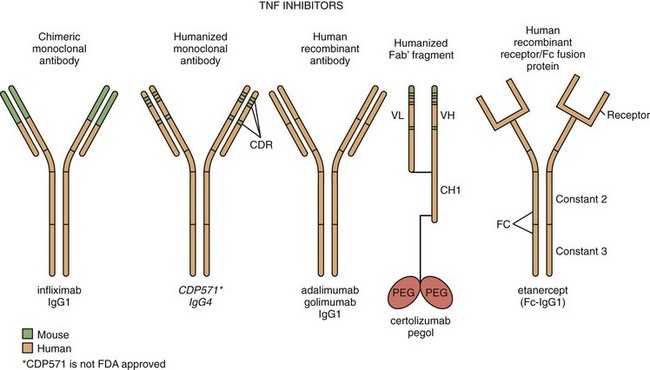
Infliximab is a chimeric mouse-human monoclonal antibody composed of constant regions of human immunoglobulin (Ig) G1κ coupled to the variable regions of a high-affinity neutralizing murine anti–human TNF antibody. The resulting construct is approximately 70% human (Figure 63-1).
Pharmacokinetics
Clinical pharmacology studies demonstrate that infliximab has a dose-dependent pharmacokinetic profile following infusions of 1 to 20 mg/kg. In combination therapy with MTX (7.5 mg once a week), serum infliximab concentrations tend to be slightly higher than when administered alone.11 Infliximab behaves in a consistent manner across different demographic groups (including pediatric vs. adult patients) and among patients with different diseases of varied severity. The half-life of infliximab is around 8 to 9.5 days at the 3 mg/kg dose, although longer values have been reported for higher doses.12 The volume distribution of infliximab at steady state (3 to 5 L) is independent of dose, suggesting a predominantly intravascular distribution.13,14 Concomitant use of MTX results in an increase in the area under the curve of infliximab of approximately 25% to 30%.
Efficacy
Rheumatoid Arthritis
In the earliest controlled trials, the efficacy of single doses of 1, 5, 10, and 20 mg/kg of infliximab was demonstrated; however, disease activity recurred when therapy was discontinued.13,15 This, along with the growing safety record, provided the rationale for studies with longer durations of therapy. In a subsequent study, concurrent therapy with MTX, even at a relatively low dose of 7.5 mg/wk, seemed to enhance the clinical response to infliximab and to decrease its immunogenicity.11 Almost all subsequent studies in RA have used such combination therapy.
Multicenter double-blind, placebo-controlled, randomized clinical trials have evaluated the effects of multiple doses of infliximab over longer periods. In the Anti-TNF Trial in Rheumatoid Arthritis with Concomitant Therapy (ATTRACT) trial, the addition of infliximab in patients with long-standing, refractory, active disease was significantly superior to treatment with MTX alone. The results were promising: Substantial improvement in signs and symptoms of disease was noted soon after treatment and was sustained though 54 weeks of follow-up.12,16 In addition to achieving substantial efficacy, the use of infliximab was associated with significant improvement in functional status and quality of life.16 Perhaps most remarkably, patients receiving infliximab had a dramatic reduction in the progression of joint damage as assessed by radiographic change scores. The median change in the Sharp score at 1 year for infliximab-treated patients was 0.0 unit (mean change, +0.55; baseline score, 50.5), indicating no significant progression. The median change in score for patients on MTX alone was +4.0 units (mean change, +7.0; baseline score, 55.5); this amount of progression is roughly what would have been predicted given the disease severity.14,16
Following the success achieved in patients with long-standing RA, this therapy was tested in patients with early RA (<3 years’ duration). In the ASPIRE trial, significant improvement in disease activity was noted in patients treated with infliximab plus MTX in comparison with MTX alone, at 54 weeks. In addition, although a significant increase in radiographic destruction was observed in patients treated only with MTX, a reduction in disease progression was observed in the infliximab plus MTX groups.17 A detailed subanalysis of data from the ATTRACT study demonstrated that a significant radiographic benefit was achieved with infliximab plus MTX treatment even among patients who experienced no improvement in signs and symptoms of disease. This suggests that there may be an uncoupling among various outcomes, and that treatment with TNF inhibitors might lead to disease-modifying activity even in the absence of a clinical response.18
In a safety-based study, a group of patients with active RA and disease more typical of clinic populations (i.e., patients with comorbidities were allowed to enroll) were treated with 3 or 10 mg/kg of infliximab for 46 weeks, resulting in comparable efficacy to that found in earlier trials.19 However, an increase in serious infectious events was reported in the 10 mg/kg group.
Psoriatic Arthritis (PsA)
In the Infliximab Multinational Psoriatic Arthritis Controlled Trial 1 (IMPACT 1), patients with or without background DMARDs were randomized to receive infliximab or placebo for 16 weeks; thereafter, all patients received infliximab until week 50. The response to infliximab therapy defined by American College of Rheumatology 20% response criteria (ACR20) and PsA response criteria was evident as early as week 2, and improvement continued through week 50. Significant improvements were also seen in skin psoriasis and in dactylitis and enthesitis assessments.20
In the phase III (IMPACT 2) trial, clinical improvement was seen very early in the infliximab-treated groups, and the response was maintained with continued treatment through the end of the study. Significant dermatologic improvement, defined by changes in the Psoriasis Area and Severity Index (PASI), was also reported in the infliximab group.21,22 In the same group of patients, infliximab inhibited radiographic progression as early as 24 weeks. Thus, the change in the van der Heijde–Sharp score was −0.70 ± 2.53 versus 0.82 ± 2.62 at week 24 for infliximab- and placebo-treated patients, respectively. At week 54, these scores continued to improve: −0.94 versus 0.53 for infliximab and placebo crossover patients, respectively.23
In the same study, the effect of infliximab on health-related quality of life was assessed. The mean percentage improvement from baseline in the health assessment questionnaire (HAQ) was 48.6% in the infliximab group, compared with a worsening of 18.4% in the placebo group at week 14. Overall, 58.6% of the infliximab group and 19.4% of the placebo group achieved a clinically meaningful improvement in HAQ at week 14.24
Ankylosing Spondylitis
The efficacy and safety of infliximab have been assessed in several multicenter clinical trials.24–27 In the initial double-blind, placebo-controlled, randomized part of one early study, disease activity measured by the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), functional ability measured by the Bath Ankylosing Spondylitis Functional Index (BASFI), and mobility of the spine measured by the Bath Ankylosing Spondylitis Metrology Index (BASMI) significantly improved with 5 mg/kg infliximab given at weeks 0, 2, 6, and 12 compared with placebo.25 Of patients who continued to receive infliximab every 6 weeks until week 102, a significantly higher percentage achieved 50% or greater improvement in the BASDAI.26,27 At week 102, 25% of the completers were in partial remission according to the Assessment in Ankylosing Spondylitis (ASAS) criteria. The clinical efficacy and the partial remission achieved in a substantial minority of patients were maintained through 3 years of treatment. In addition, the incidence of enthesitis and anterior uveitis significantly decreased during the third year of treatment in comparison with baseline.28 This group was also analyzed for the clinical response and the time to relapse after discontinuation of anti-TNF therapy. Of note, after infliximab was stopped, all patients experienced a return of disease activity, with a mean time to relapse of 17.5 weeks. Retreatment with infliximab was safe and resulted in clinical improvement in all patients to a state similar to that achieved previously.29
In another trial, most of the patients who received 5 mg/kg infliximab at 0, 2, 6, and 12 weeks reached ASAS 20% response criteria (ASAS20) at week 24, in comparison with 19% of the placebo group. Efficacy was noted as early as 2 weeks after the beginning of therapy and was maintained throughout the 24-week observation period. At week 24, 22.4% of patients in the infliximab group achieved ASAS partial remission, in comparison with 1.3% in the placebo group. In addition, patients receiving infliximab showed significant improvement in BASDAI, BASFI, and BASMI.30
To assess the effects of treatment on inflammation and structural damage, magnetic resonance images of the lumbar spine and sacroiliac joints were assessed. Spinal inflammation and clinical disease activity improved significantly from week 0 to week 30 in patients treated with infliximab plus MTX, in comparison with MTX alone.31 In one study, 2 years of continuous infliximab therapy resulted in persistent improvement in spinal inflammation determined by both T1-weighted gadolinium-enhanced and short tau inversion recovery magnetic resonance imaging (MRI) sequences in all ankylosing spondylitis (AS) patients (n = 20).32 Of note, disease activity parameters did not directly correlate with MRI results.
Etanercept
Structure
Etanercept is formed by the linkage of two soluble p75 TNF-R extracellular domains to the Fc portion of human IgG1 (see Figure 63-1). The resultant molecule binds both TNF and LT-α with high affinity and specificity.33 The TNF-R domains in etanercept bind to two of the three receptor binding sites on the TNF trimer, thus blocking the ability of TNF to interact with cell-bound TNF-R—a prerequisite for signal transduction32 (see Figure 63-1).
Pharmacokinetics
When administered subcutaneously, etanercept is absorbed slowly, reaching a mean peak concentration approximately 50 hours after a single 25-mg dose. The Ig structure affords a half-life of 3 to 4.8 days. The volume of distribution suggests predominantly intravascular distribution.34 The route of clearance from the circulation is unclear, although it is presumed to be mediated through Fc binding by the reticuloendothelial system. Different from what has been seen with anti-TNF monoclonal antibodies, concomitant use of MTX does not appear to alter the pK of etanercept.
Efficacy
Rheumatoid Arthritis
Initial studies demonstrated the efficacy and tolerability of etanercept in both early and refractory disease and also established the optimal dose as 25 mg twice weekly.35–45 In addition to achieving substantial efficacy, as measured by ACR20 clinical response criteria, the use of etanercept has been associated with significant improvement in functional status and quality of life. In one double-blind, placebo-controlled, randomized clinical trial, patients with active and long-standing RA who were refractory to DMARD therapy were treated with etanercept (10 or 25 mg twice weekly) for 6 months. Etanercept rapidly reduced disease activity.37 In another trial, the addition of etanercept in patients with active disease, despite concurrent MTX, was significantly superior to treatment with MTX alone.38 At 6 months, disease activity was significantly reduced in the combination therapy group versus those who received only MTX. In the open-label extension part of this study, patients were able to sustain the improvement, and most were able to decrease their use of MTX, corticosteroid, or both.
Following the success achieved in patients with refractory disease, the role of TNF inhibitors in the treatment of early disease was demonstrated in a large clinical trial in which two doses of etanercept (10 or 25 mg twice weekly) were compared with an accelerated dosing of MTX in MTX-naïve RA patients with disease of less than 3 years’ duration.39,40 Clinical efficacy was achieved more quickly with the standard dose of etanercept than with MTX or lower-dose etanercept. Moreover, radiographic assessments at 0, 6, 12, and 24 months showed that the rate of radiographic progression was significantly reduced with etanercept compared with MTX.
In long-term, open-label follow-up studies of patients from the clinical trials, responses to etanercept therapy have been sustained over a number of years, with some patients having received more than a decade of treatment.41 Etanercept has also proved efficacious in patients with juvenile arthritis.42 In a study known as TEMPO, etanercept plus MTX was shown to be superior to MTX or etanercept alone in patients with relatively early disease. Similarly, improvement in disability based on HAQ was greater with combination therapy. The etanercept plus MTX group showed significantly less radiographic progression than did either group receiving monotherapy, and radiographic progression was significantly less in the etanercept group compared with the MTX group. Because no pK interaction occurs between etanercept and MTX, this study conclusively shows an additive effect on various important outcome results with the combination of MTX and a TNF inhibitor.43
Different dosing of etanercept has been assessed in clinical trials. In RA patients, 50 mg etanercept once weekly resulted in similar efficacy to 25 mg twice weekly.44 Of note, etanercept monotherapy at a dose of 50 mg twice weekly did not result in increased efficacy in RA patients compared with 25 mg twice weekly.45
Psoriatic Arthritis
In the initial double-blind, placebo-controlled clinical trial, substantial improvements were observed in PsA response criteria, ACR20 response criteria, and PASI dermatologic scores in the etanercept-treated patients. The median PASI improvement was 46% in the etanercept group versus 9% in the placebo group.46
Subsequently, 205 patients with PsA were randomized to treatment with placebo or 25 mg etanercept twice weekly. At week 12, 59% of etanercept-treated patients met the ACR20 criteria, compared with 15% of placebo patients. During the open-label extension through week 48, patients continuing with etanercept treatment maintained or improved their clinical responses, while those in the placebo group showed similar improvements once they began receiving etanercept. The primary radiographic end point was the annualized rate of change in the modified total Sharp score. Radiographic disease progression was inhibited in the etanercept group (−0.03 unit) compared with worsening (+1.00 unit) in the placebo group.47
Ankylosing Spondylitis
Similarly positive results were observed with etanercept therapy in patients with ankylosing spondylitis. In a double-blind, placebo-controlled, randomized clinical trial, significant improvement in clinical response (defined by ASAS20) was demonstrated in etanercept-treated patients throughout the 24-week study.48 Sustained clinical response was maintained in the open-label extension of this study throughout 96 weeks of treatment.49 Health-related quality of life assessments also significantly improved with etanercept.50 The extent of clinical response was similar in subsequent clinical trials. Improvement measured by ASAS20 was noted as early as 2 weeks after etanercept was started, and the response was maintained throughout the trial.51 In a subsequent multicenter trial, significant improvements in BASDAI, BASFI, and BASMI scores were achieved initially in the etanercept-treated group, and later in all patients after the placebo group received etanercept as well. Relapses occurred at a mean of 6 weeks after cessation of etanercept.52 Re-administration of etanercept led to similar results in the same group of patients. Most patients were able to completely discontinue nonsteroidal anti-inflammatory drugs (NSAIDs).53
In one clinical trial, magnetic resonance images of the lower thoracic and lumbar spine of 40 patients were evaluated. Spinal inflammation regressed by 54% in the etanercept group but worsened by 13% in the placebo group after 12 weeks of therapy. After switching to etanercept, placebo patients experienced a similar improvement in spinal inflammation.54 Finally, it was demonstrated that continuous treatment with etanercept for 24 weeks reduced active spinal changes by 69% as measured by different MRI sequences.55
Adalimumab
Structure
Adalimumab is a human anti-TNF IgG1κ monoclonal antibody generated through repertoire cloning. Adalimumab neutralizes the biologic activity of TNF by binding with high affinity to the soluble and transmembrane forms of TNF and inhibiting the binding of TNF with its receptors (see Figure 63-1).
Efficacy
Rheumatoid Arthritis
In a phase II trial, adalimumab (20, 40, or 80 mg) via weekly subcutaneous injection for 12 months demonstrated efficacy in comparison with placebo.56 In a subsequent trial, adalimumab provided significant, rapid, and sustained improvement in disease activity over 24 weeks compared with MTX alone in patients with active RA despite long-term MTX therapy.57 In a study known as ARMADA, combined treatment with adalimumab and MTX demonstrated sustained improvement in the signs and symptoms of RA as assessed by American College of Rheumatology (ACR) criteria, as well as notable improvement in functional status as assessed by HAQ scores.58 In a phase III multicenter trial, 619 patients with active RA and inadequate response to MTX were randomized to receive adalimumab 40 mg every other week, adalimumab 20 mg weekly, or placebo.59 Both adalimumab regimens were significantly more effective at reducing signs and symptoms and improving physical function in comparison with placebo. In addition, joint damage was assessed radiographically using Sharp scores; patients treated with adalimumab showed significantly smaller changes, and significantly fewer adalimumab-treated patients had new erosions and overall progression of damage compared with those taking placebo. A study evaluating health-related quality of life in two clinical trials demonstrated that adalimumab plus MTX provides statistically significant improvements.60 Long-term follow-up of patients receiving open-label treatment after several controlled trials has demonstrated sustained improvement and good tolerability over several years.61
Following the efficacy noted in patients with refractory RA, adalimumab was assessed in patients with early disease. In a study known as PREMIER, adalimumab plus MTX was superior to adalimumab or MTX monotherapy in MTX-naïve patients with early RA (active disease <3 years’ duration) at 1 year. Significantly less radiographic progression was observed among patients in the combination group at both 1 year and 2 years in comparison with the monotherapy groups, although progression was less with adalimumab monotherapy than with MTX monotherapy.62 Because this study was the first to include all three possible treatment arms (TNF inhibitor, MTX, and TNF inhibitor plus MTX), it definitively established that combination therapy with MTX and TNF inhibitor achieves the best outcomes in patients with early RA. Although TNF inhibitor monotherapy was superior to MTX alone in terms of radiographic progression, clinical efficacy was comparable; however, each of these was substantially less effective than the combination.
Psoriatic Arthritis
Adalimumab given as 40-mg subcutaneous injections every other week has also been studied in patients with PsA. In a large placebo-controlled trial with 313 patients, 50% of patients were on background MTX. A significantly higher percentage of patients from the adalimumab-treated group reached ACR20, -50, and -70 response criteria compared with those receiving placebo.63 Moreover, the rate of radiographic damage progression was significantly attenuated with adalimumab. As was true in studies of the other TNF inhibitors in PsA MTX was permitted but not required for enrollment in the study. With this design, there seemed to be no difference in efficacy or toxicity between the combination of MTX plus adalimumab and adalimumab monotherapy. In addition to improvements in various articular manifestations, dramatic improvements were noted in skin psoriasis with adalimumab treatment.
Ankylosing Spondylitis
In the adalimumab trial evaluating long-term efficacy and safety in ankylosing spondylitis (known as the ATLAS trial), a total of 315 patients received either 40 mg adalimumab or placebo every other week for 24 weeks. The number of subjects who met ASAS partial remission criteria was significantly higher in the adalimumab group at weeks 12 and 24 in comparison with the placebo group. Subjects meeting ASAS 5/6 criteria (20% improvement in five of six domains, without 20% worsening in the sixth domain) at weeks 12 and 24 were also significantly more in the adalimumab-treated group.64 Significant improvement in health-related quality of life was reported in this group of patients.65 The efficacy of these drugs was demonstrated not only by improved clinical indices but also by spinal inflammation as assessed by MRI.
Golimumab
Structure
Golimumab is a human IgG1κ monoclonal antibody specific for human TNF. Golimumab was created using genetically engineered mice immunized with human TNF, resulting in an antibody with human-derived antibody variable and constant regions (see Figure 63-1).
Pharmacokinetics
Following subcutaneous administration of golimumab to healthy subjects and patients with RA, the median time to reach time to peak concentration (Tmax) ranged from 2 to 6 days. Golimumab exhibited dose-proportional pharmacokinetics in patients with active RA over the dose range of 0.1 to 10.0 mg/kg following a single intravenous dose. The volume distribution for golimumab indicates that golimumab is distributed primarily in the circulatory system with limited extravascular distribution. Median terminal half-life values were estimated to be approximately 2 weeks in healthy subjects and patients with RA, PsA, or AS. When golimumab 50 mg was administered subcutaneously to patients with RA, PsA, or AS every 4 weeks, serum concentrations appeared to reach steady state by week 12. With concomitant use of MTX, treatment with golimumab every 4 weeks resulted in a mean steady-state trough serum concentration of approximately 0.4 to 0.8 µg/mL in patients with RA, PsA, or AS. Concomitant MTX usage resulted in higher mean steady-state trough concentrations of golimumab in patients with RA, PsA, or AS (52%, 36%, and 21% respectively), compared with those treated with golimumab alone.66
Efficacy
Rheumatoid Arthritis
Results of a phase II study of golimumab in 172 patients with active RA despite MTX therapy demonstrated the efficacy of golimumab given every 4 weeks by subcutaneous injection in combination with MTX. The clinical effect was evident as early as 2 weeks after the first dose and was sustained to 1 year.67 The efficacy and safety of golimumab were evaluated in three multicenter, double-blind, placebo-controlled trials. Golimumab was administered subcutaneously at doses of 50 mg or 100 mg every 4 weeks. In the GO-FORWARD study, patients with active RA despite a stable dose of MTX who had not been previously treated with a biologic TNF inhibitor were randomized to receive golimumab 50 mg + MTX, golimumab 100 mg + MTX, or golimumab 100 mg monotherapy every 4 weeks. All golimumab regimens were significantly more effective at reducing signs and symptoms and in improving physical function.68 Patients who had less than 20% improvement in swollen and tender joint count entered early escape at week 16. The proportion of patients who achieved ACR20 and reached a low disease activity score at week 52 was significantly greater in the golimumab group. Patients who were treated with 100 mg of tocilizumab appeared to have increased risk of serious adverse events and serious infections.69
Patients who were previously treated with one or more doses of TNF inhibitor without a serious adverse reaction were enrolled in the GO-AFTER study. Greater efficacy measured by ACR20 and Disease Activity Score (DAS) 28 was observed in patients who received golimumab 50 mg plus MTX at week 14 compared with patients who received MTX alone. Improvements in HAQ scores were also markedly higher for patients receiving golimumab plus MTX in comparison with MTX alone.70
Patients with active RA who were MTX-naïve and had not been previously treated with TNF inhibitor were randomized to be treated with MTX alone, golimumab 50 mg plus MTX, golimumab 100 mg plus MTX, or golimumab 100 mg alone in the GO-BEFORE study. Golimumab plus MTX led to significant improvement in ACR responses (ACR50, 40%) at 24 weeks in MTX-naïve patients compared with patients who received MTX alone (ACR50, 29% ).71
An MRI substudy of the GO-BEFORE and GO-FORWARD trials demonstrated that patients who received golimumab plus MTX had improvements in inflammation (synovitis and osteitis) exceeding those observed with MTX only as early as week 12 and continuing through week 24. Significant improvements in erosions were also observed in the GO-BEFORE trial.72,73
Psoriatic Arthritis
The GO-REVEAL study evaluated the efficacy and safety of golimumab in PsA patients. Four hundred five patients with active PsA despite NSAID or DMARD therapy who had not been previously treated with TNF inhibitor were randomized to receive golimumab 50 mg or 100 mg with or without MTX every 4 weeks. Fifty-one percent of patients who received golimumab reached ACR20 response at week 14 in comparison with 9% of patients who received only MTX. Significant improvements in enthesitis and HAQ scores were seen in patients treated with golimumab, in addition to significant improvements in the psoriasis skin lesions evaluated with PASI responses.74 Long-term radiologic outcome was assessed using the van der Heijde–Sharp (vdH-S) method modified for PsA. Golimumab 50 mg and 100 mg given to patients with active PsA showed no to minimal evidence of radiographic disease progression through week 104.75
Ankylosing Spondylitis
The efficacy of golimumab in AS was evaluated in the GO-RAISE study. Three-hundred fifty-six patients with active AS according to modified New York criteria were randomized to receive placebo, golimumab 50 mg, or golimumab 100 mg every 4 weeks. Patients were allowed to be on stable doses of sulfasalazine, hydroxychloroquine, low-dose corticosteroids, or NSAIDs during the trial. The use of other DMARDs was prohibited. At weeks 14 and 24, golimumab 50 mg once a month demonstrated significant ASAS20 and ASAS40 responses (56% and 44%, respectively) versus placebo (23% and 15%, respectively).76
Certolizumab Pegol
Efficacy
Intravenous ascending-dose monotherapy with certolizumab pegol was shown to effectively control the signs and symptoms of RA in a phase II trial.77 The efficacy and safety of certolizumab pegol were assessed in multiple randomized, placebo-controlled, double-blind studies. RAPID-I and RAPID-II studies included patients with active RA despite MTX. Both were 52 week studies that evaluated the lyophilized (RAPID-I) or the liquid (RAPID-II) preparation of certolizumab with endpoints of progression of structural damage (change from baseline in modified Sharp score) and signs and symptoms of RA measured by ACR20, -50, and -70. Patients were randomized to receive loading doses of 400 mg of certolizumab pegol or placebo at weeks 0, 2, and 4 followed by 200 mg of certolizumab pegol, 400 mg certolizumab pegol, or placebo every other week in combination with MTX. Certolizumab pegol–treated groups showed rapid and sustained improvement in clinical disease activity starting from week 1 and sustained through week 52. Mean radiologic progression was significantly reduced in the certolizumab-treated groups versus MTX alone.78,79 Rapid and sustained improvements in health-related quality of life, fatigue, home and workplace productivity, and social activities were also reported in patients treated with certolizumab pegol in RAPID-I and -II trials.80,81 Sustainability of improvements in RA signs and symptoms and inhibition of joint damage progression, as well as tolerability of certolizumab pegol + MTX, were evaluated in the RAPID-II open-label extension study. Results at 3 years showed that 400 mg every other week dosing did not significantly increase the efficacy outcome; certolizumab pegol was generally well tolerated; and clinical and radiologic improvements were sustained over this period of time.82 In a broader group of RA patients with inadequate response to more than one DMARD, including prior TNF inhibitor exposure, addition of certolizumab pegol to the regimen resulted in rapid clinical response measured with ACR20 response rates and DAS28 scores, and improved function measured with HAQ scores.83
Mechanism of Action of TNF Inhibitors
Several potential mechanisms of action may explain the efficacy of TNF inhibitors in RA and other conditions (Table 63-1). Although some support has been put forth for these varied mechanisms, the exact relationship between any particular mechanism and specific aspects of clinical efficacy remains to be delineated. Downregulation of local and systemic proinflammatory cytokine production and reduction of lymphocyte activation and migration into the joint may be the most relevant mechanisms; for example, serum levels of IL-6 and IL-1 are significantly reduced after administration of anti-TNF monoclonal antibody.84,85 The reduction in TNF and the consequent reduction in IL-1 would be expected to reduce the synthesis of matrix metalloproteinase (MMP) and the production of other degradative enzymes. Serial studies have shown that there is in fact a marked reduction in pro–MMP-3 and pro–MMP-1 after anti-TNF therapy.86–88 As noted, anti-TNF therapy is also associated with a reduction of lymphocyte migration into the joints of patients with RA. Using radiolabeled granulocytes, it has been demonstrated that anti-TNF monoclonal antibody significantly reduces cell movement into the affected joints.89 In addition, post-treatment synovial biopsies show reduced cellular infiltrates, with fewer T cells and macrophages present.90 These effects are thought to be secondary to a reduction in the expression of endothelial adhesion molecules in the synovial tissue. Treatment with anti-TNF monoclonal antibody results in a dose-dependent decrease in soluble forms of intercellular adhesion molecule-1 (ICAM-1) and E-selectin (CD62E).89 Changes in soluble E-selectin, soluble ICAM-1, and circulating lymphocytes with anti-TNF therapy correlate with clinical outcomes. Vascular endothelial growth factor (VEGF) is a potent endothelial cell–specific angiogenic factor. It is produced in the synovium and is an important regulator of neovascularization in the pannus. After anti-TNF therapy, VEGF serum levels are reduced in patients with RA. This decrease correlates significantly with the clinical benefit observed in these patients.91 Because angiogenesis is a prominent feature of rheumatoid synovium, the relationship between inflammation and angiogenesis has been investigated. Computerized image analysis of endothelium for multiple markers of endothelium (e.g., von Willebrand factor, CD31) and neovasculature (αvβ3) has shown reduced vascularity after anti-TNF therapy. A number of other potential mechanisms of action have been suggested to be operative for TNF inhibitors (see Table 63-1), although debate on these aspects is ongoing.
Table 63-1 Potential Mechanisms of Action of Tumor Necrosis Factor Inhibitors
| Decrease Production of Other Inflammatory Mediators |
| Alter Vascular Function; Leukocyte Traffic and Activation |
| Modulate the Function of Immunocompetent Cells |
| T Cells |
| Monocytes and Macrophages |
GM-CSF, granulocyte-macrophage colony-stimulating factor; HLA-DR, human leukocyte antigen DR; IL, interleukin; MMPs, matrix metalloproteinases; Th, T helper.
Other Considerations
Treatment in Other Autoimmune Conditions
The role of TNF inhibitors in the treatment of Crohn’s disease, juvenile idiopathic arthritis, and psoriasis has been clearly defined. Based on promising results in various immune conditions, these agents have been used in a variety of other disorders, including idiopathic and spondyloarthopathy-related anterior uveitis, sarcoidosis, Sjögren’s syndrome, Behçet’s syndrome, inflammatory myopathies, and various types of vasculitis. Although a number of case reports or small, uncontrolled clinical trials have reported on these conditions, there is a paucity of conclusive data from controlled trials. Perhaps the most notable clinical response has been observed in the treatment of anterior uveitis, especially with anti-TNF monoclonal antibody constructs.92 On occasion, promising results in uncontrolled trials have been disproved in controlled trials. This was observed in a placebo-controlled trial of etanercept given in addition to standard therapy for remission induction and maintenance in patients with granulomatosis with polyangiitis (formerly Wegener’s granulomatosis). Despite promising anecdotal evidence, the addition of the TNF inhibitor not only failed to achieve any clinical improvement but also resulted in greater risk of solid malignancies beyond that observed with cyclophosphamide alone.93
Despite evidence that anti-TNF agents can result in the development of certain autoantibodies and even lupus-like syndromes, the safety and efficacy of anti-TNF agents have been assessed in a small group of patients with systemic lupus erythematosus (SLE). Patients with joint involvement experienced remission of arthritis, and a significant reduction in the level of proteinuria occurred with infliximab. In this small study, TNF inhibitor therapy did not lead to adverse events suggestive of an increase in SLE activity; however, as might have been expected, autoantibodies to double-stranded DNA and cardiolipin did increase.94
Cardiovascular Risk and Lipid Profile
Cardiovascular morbidity and mortality appear to be increased in autoimmune conditions. This may be related both to increased prevalence of traditional risk factors for cardiovascular disease and to uncontrolled systemic inflammation, which appears to predispose independently to accelerated progression of atherosclerosis. Treatment with TNF inhibitors appears to be associated with overall improvement in cardiovascular disease risk, related to beneficial effects on lipid parameters and to control of systemic inflammation.95,96 Long-term investigations are needed to define the possible beneficial effects of TNF inhibitors on overall and cardiovascular survival in patients with autoimmune disease.95,96
Pregnancy and Breastfeeding
Developmental toxicity studies in rats, rabbits, and mice have not revealed any maternal toxicity, embryo toxicity, or teratogenicity associated with TNF inhibition. Minimal human pregnancy information has been published for these medications, and most data consist of isolated case reports, retrospective surveys, and uncontrolled studies. As the number of patients treated with TNF inhibitors increases, a growing number of pregnancies will be reported among them.97 Outcome data based on anecdotal observations of small numbers of pregnant women treated with infliximab, etanercept, and adalimumab reveal that the relative rates of live births, miscarriages, and therapeutic terminations were comparable with rates in a national cohort of age-matched healthy women. TNF inhibitors are classified as U.S. Food and Drug Administration (FDA) Pregnancy Category B (animal reproduction studies have failed to demonstrate a risk to the fetus, and no adequate and well-controlled studies have been performed in pregnant women). The use of anti-TNF agents in pregnancy is recommended only if such treatment is clearly needed. If TNF inhibitors are used during pregnancy, it must be noted that transfer to the fetus is possible, and monitoring might be considered on that basis. Because it is not currently known whether TNF blockers are excreted in human milk, or whether they are absorbed systemically after ingestion, it is recommended that TNF blockers not be used by nursing mothers.
Toxicity
In clinical trials, etanercept, infliximab, adalimumab, golimumab, and certolizumab pegol have generally been well tolerated.* Longer-term follow-up of patients initially enrolled in clinical trials has provided additional safety data for these agents. However, TNF plays a key role not only in the pathogenesis of autoimmune disease but also in normal immune homeostasis. Therefore, a number of safety considerations, including the potential risk of infection and malignancy, are germane to the optimal clinical use of these agents.98
Additional information concerning adverse effects associated with these agents has been obtained through pharmacovigilance. Adverse events related to the use of TNF inhibitors can be grouped into those that are agent-related and those that are target-related (Table 63-2).99 Injection site and infusion reactions and immunogenicity and their sequelae vary, depending on the particular agent. A potentially increased predisposition to infection, development of malignancy, and induction of autoimmune disorders and an association with demyelinating disorders, myelosuppression, and worse outcomes with congestive heart failure might be considered target-related adverse events. Thus, any clinically effective TNF inhibitor might be expected to be associated with such adverse events, although the relative risk among different agents may vary, depending on dose and other factors.
Table 63-2 Adverse Effects Potentially Associated with Tumor Necrosis Factor Inhibitors
| Target-related |
| Agent-related |
Infusion and Injection Site Reactions
Infliximab has been associated with infusion reactions, the most common of which are headache (20%) and nausea (15%). These are rarely severe, are usually transient, and typically can be controlled by slowing the rate of infusion or by treating with acetaminophen or antihistamines.12,16 With etanercept, adalimumab, golimumab, and certolizumab pegol, cutaneous reactions at injection sites represent the most frequent administration-related side effect; however, they rarely lead to discontinuation of therapy.30,58,70,81 Injection site reactions typically consist of erythematous or urticarial lesions. Although they can arise at sites of previous injections, these reactions seem to be limited to the skin and are not associated with other or systemic features of immediate hypersensitivity. Reactions typically occur close to treatment initiation and abate over time, even with continued dosing.
Antigenicity
As is true for any therapeutic agent (especially large protein molecules, some of which contain foreign sequences), antibodies to anti-TNF agents can develop. Although the clinical relevance of these antibodies is presently unclear, they can diminish the half-life of the therapeutic agent and consequently decrease its efficacy. Approximately 3% of etanercept-treated patients develop antibodies to the drug. In an early study, it was noted that antibodies to infliximab developed in 53%, 21%, and 7% of patients who were receiving 10, 3, and 1 mg/kg infliximab, respectively.12 RA trials of infliximab with or without concomitant MTX treatment revealed that immunogenicity was decreased by concomitant MTX, perhaps owing in part to the increase in the half-life of infliximab associated with MTX use.12 A multicenter trial of infliximab therapy in Crohn’s disease demonstrated that induction of these anti-infliximab antibodies might contribute to hypersensitivity reactions in some patients. Antibodies to adalimumab, golimumab, and certolizumab pegol developed in about 4% to 12% of patients; this rate was reduced to 1% with concurrent MTX treatment.70–76,78–81,99 Although it is believed that there is a trend toward higher clearance of TNF inhibitors in the presence of antibodies to the construct, routine testing for antibodies to TNF inhibitors is not widely available, nor is it currently recommended.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


