106 Etiology and Pathogenesis of Juvenile Idiopathic Arthritis
Juvenile idiopathic arthritis (JIA) comprises a heterogeneous group of diseases that lead to a final common pathway, namely thickening and inflammation of the joint lining with characteristic onset in children. Infiltrating inflammatory cells interact with each other and resident synovial fibroblasts, promoting a chronic inflammatory process in the synovial membrane and secreted synovial fluid (Figure 106-1). In this chapter, we discuss the genetic factors that predispose to JIA and the abnormalities that underlie synovial and systemic inflammation. In addition to the established association between the major histocompatibility complex (MHC) loci and JIA, more recent studies have detected links to genes that regulate cellular activation or cytokine responses.1 The fact that immune-related genes make up the major risk alleles in JIA strongly supports the concept of JIA as a disease of disordered immunity. Highly activated T cells, monocytes, and neutrophils are attracted to joint and secrete mediators that perpetuate inflammation and also attenuate immune regulation. The relative importance of individual cytokines varies between disease subtypes. Results from therapeutic trials support a role for tumor necrosis factor (TNF) in the pathology of polyarthritis forms of JIA and for interleukin (IL)-1β and IL-6 in systemic JIA (sJIA) (Table 106-1). The past decade has seen a step change in the quality of treatments in JIA. To build on this success, researchers need to be able to interrogate the vast array of biologic data that is emerging in the field of JIA and translate this knowledge into novel therapies and a more tailored treatment approach for our patients.
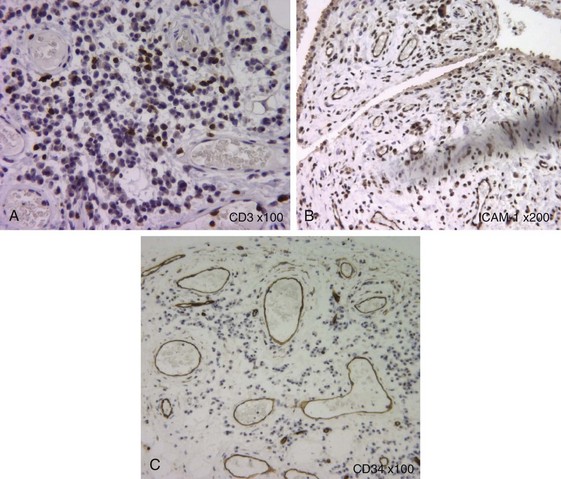
Table 106-1 Summary of Cytokines and Inflammatory Mediators Implicated in Juvenile Idiopathic Arthritis
| Cytokine/Mediator | Cell | Pathology |
|---|---|---|
| TNF | Monocytes, T, B cells, PMN, mast cells, fibroblasts | |
| IL-1β | Monocytes, B cells, fibroblasts | |
| IL-17 | T cells (Th17), mast cells | |
| IL-6 | Monocytes, fibroblasts, B cells | |
| IFN-γ | T cells (CD4+ Th1, CD8+, NK cells) | |
| MRP 8/14 | Monocytes, PMN |
IFN-γ, interferon-gamma; IL, interleukin; MRP, myeloid related protein; NK, natural killer; PMN, polymorphonuclear neutrophil; TNF, tumor necrosis factor.
The term juvenile idiopathic arthritis refers to a group of conditions, defined under the International League of Associations for Rheumatology (ILAR) classification as conditions starting before the sixteenth birthday that are characterized by arthritis of at least one joint that persists for 6 weeks or more, as a common feature.2 Although this classification has proven highly valuable for both clinical and basic research and enables more precisely defined subgroups and comparison of data from different studies, it does not encompass all aspects of the heterogeneity of childhood arthritis (e.g., use of the classification criteria by strict exclusion rules may lead to up to 30% of cases being designated as “unclassified”). In addition, certain common features may occur across several subtypes (e.g., factors associated with risk of autoimmune uveitis of JIA include positivity of the antinuclear antibody [ANA], early age at onset, and female sex). Girls with young-age onset of arthritis and ANA positivity represent a group of patients who have been proposed to represent a relatively homogeneous group, yet they currently fall into several subtypes within the ILAR classification. Similarly, genes and immunologic processes that influence the likelihood of mild oligoarthritis to extend to more severe arthritis may well overlap with etiopathologic factors involved in polyarticular JIA.3,4 Thus in the future a full molecular and genetic analysis of the heterogeneity of childhood arthritis may permit the development of a more mechanism-driven classification, which could help inform on likely disease course, complications, or response to treatment.
Histologic Features of Juvenile Idiopathic Arthritis Inflamed Synovium
The pathologic hallmark of juvenile idiopathic arthritis is the inflamed synovium. Histology of this tissue shows thickened synovium that is highly vascular and shows marked hyperplasia of synoviocytes in the lining layer, as well as a dense infiltrate of inflammatory cells, comprising T cells, macrophages, dendritic cells, and in some cases B cells and natural killer (NK) cells5,6 (see Figure 106-1). The hypertrophied synovial layer is highly vascular, with endothelium expressing markers of activation such as human leukocyte antigen (HLA)-DR and intracellular adhesion molecule 1 (ICAM-1). The vascularity is likely related to the increased production of proangiogenic factors such as vascular endothelial growth factor (VEGF), osteopontin, and the angiogenic chemokines,7–9 while recruitment of this inflammatory infiltrate is likely mediated by multiple chemokines shown to be increased in JIA including CCL5, CXCL10, CCL20, IL-8, and MCP-1 among others.10,11 The proteolytic enzymes matrix metalloproteinases MMP-1 and MMP-3 are abundantly expressed in synovial lining, and their levels correlate with degree of infiltration, in particular by myeloid CD68+ cells.11 The T cells that infiltrate the inflamed synovium, like those in the excess synovial fluid, are highly activated memory cells12–14 of both CD4 and CD8 populations.15–17
Genetics of Juvenile Idiopathic Arthritis
Key Points
There is strong evidence for a genetic component to the etiology of JIA.
The strongest genetic associations of JIA are with genes of the MHC.
Specific MHC class I alleles are associated with enthesitis-related arthritis (ERA).
Distinct MHC class II alleles are associated with oligoarthritis and polyarthritis subtypes.
There is strong evidence for a genetic component to the etiology of the JIA diseases. Twin studies have shown concordance rates in monozygotic twins of between 20% and 40%.18 A study of 164 affected sibling pairs (ASPs) with JIA showed a 70% concordance for gender, 73% for age at disease onset, and 66% for disease course.19 Estimates suggest that the recurrence risk for siblings of a proband with JIA are between 15 and 30 times that of the general pediatric population, a figure that is as high as for insulin-dependent diabetes mellitus (IDDM) or multiple sclerosis.20 In addition, first-degree relatives of children with JIA have a higher rate of other autoimmune disease than controls.21 Despite this evidence, families of multiple affected siblings are still relatively rare. One caveat in comparing early studies is that some were performed using the American College of Rheumatology (ACR) classification criteria (at that time called juvenile rheumatoid arthritis), so they may not be directly comparable with studies that have used the current ILAR classification of JIA. However, there is now a growing body of data that has been analyzed using the ILAR criteria, making comparisons feasible.
Genetic influences on JIA susceptibility and phenotype are polygenic, such that JIA is thought of as a complex genetic trait.20 Although some studies have analyzed all forms of JIA together, the heterogeneity of childhood arthritis would lead to the prediction of different genetic risk associations for different disease phenotypes, and this has been confirmed in recent adequately powered studies. The unique features of different subtypes of JIA mean that it is perhaps unsurprising that there are also genetic associations with specific subtypes.
The strongest genetic associations with JIA are of genes that lie within the MHC, or HLA; these were the first to be documented as associated with childhood arthritis.22 The MHC region, located on chromosome 6 of the human genome, includes more than 200 genes, many of which are central to functions of the immune system. Numerous associations between HLA genes and JIA have been reported, and these involve many different populations (reviewed by Prahalad and Glass1). The best characterized associations are with genes of the so-called MHC class I and class II loci, genes that code for heterodimeric proteins that present peptide antigen to the specific antigen receptor of T cells (the TCR).
Class I loci include HLA-A, HLA-B, and HLA-C, of which the earliest report was of the association between HLA-B27 and the form of JIA known as enthesitis-related arthritis (ERA), which includes children whose disease has parallels with adult ankylosing spondylitis (see Chapters 74 and 75). The HLA-A2 allele HLA-A*0201 is increased in several types of JIA, especially oligoarthritis and children with early-age onset.23,24 A recent study has also implicated HLA-C*0202 in association with persistent oligoarticular JIA.25
Multiple studies have revealed associations of class II MHC loci (HLA-DR, HLA-DP, and HLA-DQ) with JIA, of which the strongest are DRB1*0801 and 11 with oligoarticular JIA, as well as DRB1*1301, in particular in ANA-positive cases.26 Because of inheritance of genes in the MHC region together in so-called haplotypes due to linkage disequilibrium (LD), observed associations may in fact be due to genes within the haplotype, distinct from the locus first implicated. Several haplotypes across the MHC confer an increased risk for all types of JIA such as DRB1*08-DQA1*0401-DQB1*0402, which confers an odds ratio of 6.1 and 10.3 for persistent and extended oligoarticular JIA, respectively. Frequency of the DRB1*1301-DQA1*01-DQB1*06 haplotype distinguishes persistent from extended oligoarticular JIA, whereas DRB1*0801 and DRB1*1401 are associated with polyarticular JIA.26 Together, these effects may be large: In one study the presence of the combination of the HLA-DRB1*0801, HLA-DRB1*1101, and DPB1*0201 alleles conferred a relative risk of 236.27 Some associations closely mirror those of the corresponding adult disease such as the strong association of the HLA-B27 allele with spondyloarthropathy and the HLA-DRB1*0401 with rheumatoid factor (RF)-positive polyarticular JIA. Although sJIA shows less strong associations with HLA alleles, even in this subtype, specific haplotypes (such as DRB1*11-DQA1*05-DQB1*03) are increased compared with control subjects. A recent study that used fine allele-specific typing across eight HLA loci in a large cohort and analysis by haplotype has confirmed that within class II loci, HLA-DR is the driving association, rather than HLA-DP.25 Remarkably, some HLA allele/JIA subtype associations show age-specific effects, in that they confer risk over a specific age range only.24 Another recent study confirmed both age- and gender-specific effects.25 Thus, for example, in polyarticular JIA the HLA-DRB1*0801 allele has risk effect in those whose arthritis starts after the age of 6, whereas in younger-onset polyarticular JIA the risk alleles are more closely related to those of patients with oligoarticular JIA, HLAB1*1103/1104.25 In addition to these risk alleles, some alleles, notably DRB1*0401, *0701 and *1501, are consistently reduced in frequency in JIA cases compared with controls, suggesting a protective effect, for some or even all subtypes. These associations between genes that code for proteins whose central function is the presentation of antigenic peptides to T cells implicate a T cell–driven process in at least part of the etiopathogenesis of JIA.
In addition to the genes coding for MHC proteins, a large number of other candidate genes (now totaling more than 100) have been the focus in different studies of JIA.28 Inflammatory cytokines have been an important target for drug development in JIA over the past decade, and similarly their gene polymorphisms have been a key area of scrutiny. Genes studied have included cytokine and chemokine genes such as IL-1, IL-6, TNF, MIF, and IL-10; their receptors (including IL-1R, IL-2RA, and CCR5); and key signaling molecules including CTLA4 and PTPN22. However, only a few of these loci have been independently validated: PTPN22, MIF, SLC11A6, WISP3, TNF,1,29 and in a recent meta-analysis, CCR5.30
Genetic association at the TNF locus is complex to study given the position of the TNF gene within the MHC, but some data suggest that several HLA-independent TNF haplotypes are significantly associated with JIA, although the functional consequences of these alleles are not yet clear.31 The hypothesis suggesting a link between sJIA and IL-6 was proposed many years ago because several clinical features in sJIA resembled the phenotype of IL-6 overexpression (e.g., fevers, stunted growth, anemia).32,33 A polymorphism (−174G/C) in the regulatory region of the IL-6 gene alters transcription of IL-6 in response to IL-1 and LPS; sJIA patients have significantly lower frequency of the protective CC genotype,34 and the IL-6 −174G allele has been confirmed as a susceptibility gene for sJIA.35 Macrophage inhibitory factor (MIF) gene is associated with JIA.36 This polymorphism (MIF −173*C) results in higher MIF production in the serum and synovium of JIA patients and has been suggested to be predictive of outcome of intra-articular steroid injections in sJIA.37
The apparently conflicting studies on several of these candidate gene loci reflect two problems that have hindered progress: the heterogeneity of JIA, which means that combining all JIA into one study may lead to loss of detection of effects specific for a subtype, and opposing this, the difficulty of reaching adequate power for genetic studies, if stratification by subtype is preferred. Recently, new insights have come from the application of new knowledge from other diseases to the understanding of JIA genetics. Thus the highly successful Wellcome Trust case control Consortium performed genome-wide association studies in seven major common diseases including rheumatoid arthritis (RA).38 Extrapolation of new loci implicated in RA to JIA has been fruitful and has suggested that other key loci that show association with JIA are IL-2/IL-21 and IL-2RA, the α chain of the IL-2 receptor, also known as CD25.39,40 A recent study focused on specific subtypes of JIA and comparison with their adult disease counterparts. The genetic loci IL-23R and endoplasmic reticulum (ER) aminopeptidase-1 (ERAP1) have been identified as carrying risk associations with ankylosing spondylitis (AS) and psoriatic arthritis in adults.41 Genotyping of these genes in a large cohort of JIA cases has shown associations of both with the ERA subtype of JIA but no association with JIA as a whole, again emphasizing subtype-specific genetic features and likely pathogenesis.42 ERAP1 is of particular interest given its functional role in trimming of peptides within the ER, for presentation of peptide on, and folding of, MHC class I molecules, and given the strong evidence for MHC class I folding abnormalities in patients with the HLA-B27 risk alleles (HLA-B*2705 and *2702 among others41); see also section on enthesitis-related arthritis (and Chapter 74). ERAP1 is also thought to have a role in trimming of cytokine receptors at the cell surface. IL-23 is of great functional interest because of the central role of IL-23 in the Th17 pathways, as well as the demonstration that Th17 cells may play a role in both JIA and adult AS/psoriatic arthritis.
Adaptive Immune System
T Cells
The strong association of many JIA subtypes with genetic variants at HLA loci, as well as the central role of HLA proteins in presenting peptide antigens to T cells for recognition, which is central to T cell function, led to intense investigation of the role of T cells in the pathology of JIA. Highly activated memory T cells make up a significant proportion of the inflammatory infiltrate in JIA and express an “oligoclonal” or restricted set of T cell receptors (TCRs).14,16,17 Specific T cell clones can be long-lived and are detectable in different inflamed joints.17 Nevertheless, it is still unclear whether these clones represent autoreactive T cells specific for an “arthritogenic” epitope, akin to islet cell antigens in type I diabetes. Although immune activation leads to tissue damage in JIA, the role of self-antigen recognition in this process remains unclear. However, the association of JIA with genetic loci that influence the threshold of T cell activation such as PTPN22, as well as those central to recognition of antigen by T cells, supports the concept of JIA as a disease of dysregulated adaptive immunity.
Early work examining animal models of arthritis led to the hypothesis that cells of the T helper 1 (Th1) lineage, secreting interferon-γ (IFN-γ), were central to pathogenesis. Th1 cells are recruited to the joint by high levels of chemokines CCL5, macrophage inflammatory protein (MIP)-1α, and IP-1043 and make up the majority of T cells in the JIA joint.12 However, in both mouse models of arthritis and early adult clinical trials, blocking IFN-γ has offered little clinical benefit, which suggests that other players may be important to pathogenicity. TNF, the prototypic inflammatory cytokine in arthritis, is secreted by T cells and macrophages within the joint and is detectable within inflamed synovial tissue,44 and to a lesser extent in synovial fluid.45 The success of TNF blockade in polyarthritis and extended oligoarthritis subtypes implicates TNF in JIA pathology.46 Still, up to a third of patients fail to respond adequately or relapse on anti-TNF therapy and a recently discovered T cell population, “Th17” secreting IL-17 and IL-22, may account for this recalcitrant disease.47 IL-17 causes significant bony and cartilage damage in the joint by promoting neutrophil influx via the secretion of IL-8 and synergizing with IL-1β and TNF to drive metalloproteinase secretion and osteoclast activation.48 Th17 cells are enriched in the joints of JIA patients, and Th17 numbers correlate with the severity of disease course in oligoarthritis.11 Recent evidence suggests that a significant proportion of the inflammatory T cells in the joint have a Th17 ancestry, raising the hope that Th17 blockade will be an effective treatment in some subtypes of JIA.49
In addition to proinflammatory processes, there is strong evidence for ongoing immune regulation in JIA. Children with persistent oligoarticular JIA have high numbers of a regulatory subset of T cells (Treg) within the joint that express CD25 and Foxp3, and the number of Tregs is significantly higher in the children with persistent oligoarticular JIA than those with the more severe extended oligoarticular disease.50,51 In addition, T cells specific for the conserved self-antigen heat shock proteins (HSPs) play a similar regulatory role.52 Although synovial Treg suppress effector T cell functions in vitro, inflammatory cytokines such as IL-6 and TNF, present in the arthritic joint,10,53 may attenuate Treg function in vivo. Treatment strategies that expand Treg numbers and augment function are currently under investigation.54–56
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


