28 Experimental Models for Rheumatoid Arthritis
Animal models are tools to mimic various aspects of rheumatoid arthritis (RA).
Animal models provide direction for novel approaches to treatment such as cytokine inhibition.
The advantages of using animal models are mainly the following:
1. Genetically controlling inbred animal strains.
2. Controlling their environment better than for humans.
3. Manipulating experiments. Researchers can change the genome of inbred strains by mutations, insertions, and deletions. They can also change the environment in a controlled way, such as immunizing or infecting animals, which may lead to arthritis. Controlled experiments can be performed.
For animal models to be useful, they need to recapitulate some of the key features of RA, such as:
• The disease starts before the clinical diagnosis. An autoimmune and inflammatory process precedes the clinical onset by several years.
• Tissue specificity. RA is characterized by a tissue-specific inflammatory attack affecting diarthrodial, peripheral, and cartilaginous joints. Although systemic immune responses and manifestations are usually present, the inflammation is mainly directed toward peripheral joints.
• Chronicity. The disease is chronic and occurs in tissues in which no causative infectious pathogens have so far been demonstrated. Acute joint affections are common manifestations in both physiologic responses to infections and in connection with other inflammatory disorders, but in RA chronicity is an essential characteristic. The disease course may proceed with identifiable relapses, but there is usually a steady progression of joint destruction.
• Autoantibodies. The development of RA is preceded and associated with elevated levels of autoantibodies in serum. Antibodies to citrullinated protein (ACPA) have the highest specificity and sensitivity followed by antibodies to immunoglobulin (rheumatoid factors), but antibodies to other antigens also occur in subsets of patients such as antibodies to type II collagen (CII) and hnRNP-A2.
• Major histocompatibility complex (MHC) class II association. The genetic influence is significant but complex. Class II genes in the major histocompatibility complex make the largest genetic contribution by far. In particular, certain structures in the peptide-binding pocket of HLA-DR4 molecules are highly associated with RA. Several other loci confirm that involvement of adaptive immunity (PTPN22, CTLA4, IL-21) strengthens the view that RA is an autoimmune disease.
Taken together, these findings suggest that both activation of innate immunity and immune-mediated inflammation directed to peripheral joints play a role in the disease process. Because the disease process starts several years before clinical onset with enhanced levels of autoantibodies (ACPA and rheumatoid factor [RF]) together with a higher level of inflammation markers, it is likely that the etiologic factors are operating at this early time. Smoking and various chronic infections such as periodontitis have been suggested to be associated with the early disease process. This process is, however, not joint specific and a further spreading of the autoimmune reactivities toward joint specificities is likely to occur before onset of arthritis. One possible explanation for such a response is an occurrence of an infectious agent persisting in the joint such an agent has not been identified as an explanation for RA, although infectious agents have been shown to induce and promote arthritis. Alternatively, the immune reaction could be directed to molecular targets in the joints, in cartilage, or in the synovial tissue. Another explanation could relate to a defect in a gene related to peripheral joints (e.g., leading to cartilage fragility or a gene affecting immune recognition). However, such a genetic defect has not been found. Instead, genome-wide association studies suggest that the MHC class II region contains the most important genes supplemented with a large number of genes outside the MHC region, most of which are associated with adaptive immune responses. Thus the cause and driving forces are polygenic and multifactorial, and the understanding of the disease will require a detailed basic analysis of disease mechanisms. Animal models are excellent tools for this type of analysis. Recent advancement of different animal models mimicking different aspects of human diseases, as well as the improvement in genetic techniques, has dramatically increased their usefulness. The present overview includes not only models for RA but also briefly adds models with related disease pathways such as psoriasis arthritis, reactive arthritis, ankylosing spondylitis, Lyme disease, and septic arthritis (Table 28-1). However, there is a focus on the classical models for RA that are currently commonly used in both industry and academia: the adjuvant arthritis models in the rat, the collagen-induced arthritis (CIA), and the collagen antibody–induced arthritis (CAIA).
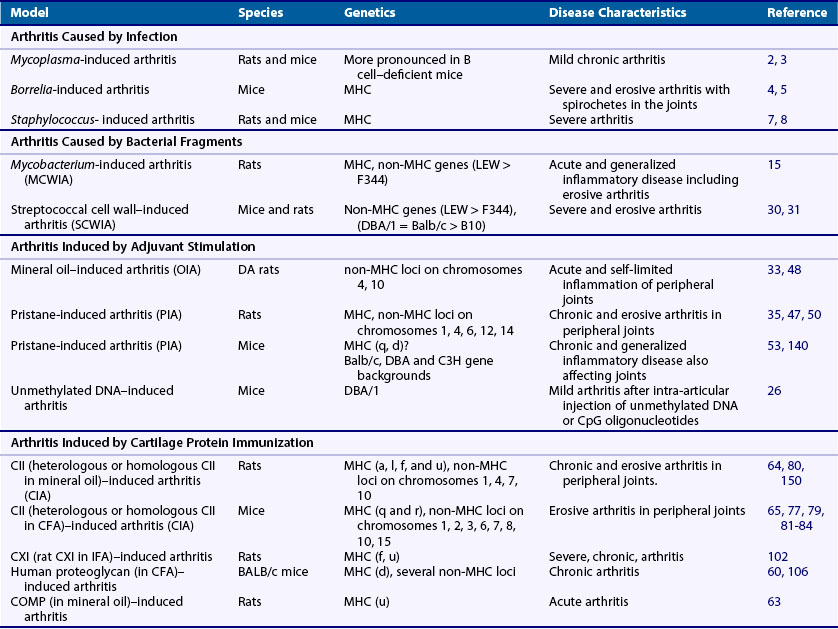
Arthritis Caused By Infectious Agents
Mycoplasma arthritides Arthritis
Arthritis associated with mycoplasma infection is endemic among farm animals. It is also possible to induce arthritis in rodents after inoculation with Mycoplasma arthritidis. However, mycoplasma bacteria are not easily found in RA joints, although they may cause arthritis in individuals with severe B cell deficiency.1 Inoculation of mice induces a mild chronic arthritis in conjunction with the persistence of the microorganism.2 In accordance with the observations in humans, B cell depleted mice are more susceptible to mycoplasma–induced arthritis.3
Lyme Arthritis
Borrelia is a spirochete that may persist in joints and cause arthritis. The clinical picture is chronic and resembles RA, and it is genetically associated with MHC class II-DR4, as is RA. Clearly, live bacteria persist in the joints but in many patients it has been difficult to identify the spirochete in the arthritic joints. Mice infected with Borrelia develop arthritis similar to the human disease.4 As in humans, MHC controls the susceptibility to arthritis and the immune response associated with human MHC class II expressed in mice has been shown to be directed to Borrelia-derived antigens.5 The persistence of the spirochete seems to be a requirement for the development of the arthritis,6 although some mouse strains do not develop arthritis in spite of high levels of bacteria in the joints.
Staphylococcal Arthritis
Septic arthritis is most commonly caused by a persistent infection of Staphylococcus aureus. The bacteria tend to be encapsulated in tissues including joint synovia and persist for years. Inoculation with certain S. aureus strains induces septic arthritis in many mouse and rat strains.7,8 Severe and prolonged arthritis develops in infected joints mimicking the human situation. Interestingly, protection of the host is critically dependent on the innate defense such as neutrophils and complement, whereas the adapted immune response is not effective.9 Instead, the apparently aberrant adapted immune response promotes arthritis.10,11
Arthritis and Ankylosing Spondylitis Induced by Intracellular Bacteria
Some bacteria with the capacity to invade cells on infection (e.g., Yersinia) are known to be related to postinfectious arthritides such as reactive arthritis and ankylosing spondylitis. These diseases are genetically associated with HLA-B27, a MHC class I allele of the B locus. It has been possible to reproduce the human disease to a large extent in HLA-B27 transgenic mice and rats.12 In B27-transgenic rats, ankylosing spondylitis, balanitis, colitis, dermatitis, and arthritis occur spontaneously. However, if the rats are made germ free, the joint manifestations are no longer present, indicating the importance of a so far unknown infectious agent.13 A similar phenomenon has been shown to occur in B27 transgenic mice,14 in which arthritis occurs only in conventional animal facilities.
Arthritis Caused by Bacterial Fragments
Postinfectious arthritis can develop after bacterial infections. The occurrence of arthritis can be dependent on several different bacteria-derived compounds such as cell wall fragments, DNA, and heat shock proteins. Bacterial cell wall fragments are difficult to degrade and may cause prolonged activation of macrophages and synovial macrophages. The first animal model for RA to be described was the so-called adjuvant arthritis (mycobacteria cell wall– induced arthritis, MCWIA) induced in rats after injection of mycobacterium cell walls suspended in mineral oil (i.e., complete Freund’s adjuvant [CFA]).15 Only rats (and not mice or primates) develop arthritis after mycobacterium challenge,16 although it has been reported that joint-related granuloma formation has occurred in humans treated with mycobacterium-containing vaccine.17 CFA is a potent adjuvant that activates a multitude of pattern recognition receptors, activating antigen-presenting cells enhancing T cell immunity. Subcutaneous injection of CFA in rats leads to granulomatous inflammation in many organs (e.g., the spleen, liver, bone marrow, skin, and eyes) and causes profound inflammation in peripheral joints.18 MCWIA is severe but self-limited, and the inflammation subsides after 5 to 7 weeks. The mycobacterium cell wall fragments are most likely disseminated throughout the body and engulfed by tissue macrophages, which have difficulties in degrading the bacterial cell wall structures and are therefore transformed into an activated state, which trigger inflammation.
MCWIA can be abrogated by elimination of the classical α/β type of T cells and spleen-derived T cells19,20 can transfer the disease. The specificity of such T cells has, however, not been reproducibly demonstrated, although some possibilities have been suggested including bacterial structures and cross-reactive self-components.21,22 Although a role for heat shock proteins as T cell antigens has not been confirmed, they play a regulatory role for the development of arthritis.23 In the search for the minimal arthritogenic structure in mycobacterium, it was observed that one of the essential structural elements of the mycobacterium peptidoglycan, muramyl dipeptide, could induce arthritis.24 Interestingly, T cells do not recognize this structure, but it has potent adjuvant capacity as it activates the inflammasome by stimulating innate immune receptors (NOD2) and antigen-presenting cells.25 The unmethylated DNA of bacteria has also been shown to independently trigger arthritis in mice26 and contribute to arthritis severity of MCWIA in rats.27 The bacterial DNA triggers Toll-like receptors on both antigen-presenting cells and inflammatory macrophages and will therefore interact with both T cell–dependent and inflammatory pathways. Another T cell–dependent arthritogenic pathway is triggered by the mineral oil in which the mycobacteria is suspended, as is discussed later in more detail.19–24 Thus this classical “adjuvant-induced arthritis” (MCWIA) is mediated by different and interacting pathways, dependent on both different mycobacterium cell components such as peptidoglycans, DNA, and heat shock proteins but also dependent on adjuvant activity mediated by the oil used to suspend the mycobacteria.
Postinfectious arthritis has also been observed to occur following streptococcal infections. A rapidly developing form of arthritis has been observed after systemic inoculation of streptococcal cell wall fragments in rats28 and mice29 but not in primates.16 Peptidoglycans from the cell wall rapidly disseminate throughout the rat including the joints.30 These structures are difficult to degrade for the macrophages, and as a consequence synovial macrophages are persistently activated. T cells are necessary for the initiation and perpetuation of the arthritis.31 Although the precise mechanisms are not known, it is possible that there are mechanisms shared with MCWIA.
Nonbacterial Adjuvant-Induced Arthritides
Research has found that the induction of arthritis in rats was dependent on not only the mycobacteria but also the oil into which the mycobacterium fragments were suspended. Interestingly, some oils supported the induction of arthritis, whereas others did not.32 Many years later it was noted that the mineral oils that supported the induction of arthritis were in fact arthritogenic by themselves.33 It was also found that subcutaneous administration of nonbacterial adjuvant compounds such as pristane, hexadecane, and squalen were highly effective in inducing arthritis.34–36 These adjuvant compounds in most cases produce inflammation confined to the joints and offer more appropriate experimental models for RA than the earlier commonly used “adjuvant arthritis.”
Mineral oil–induced arthritis (OIA),33 avridine-induced arthritis (AvIA),34 pristane-induced arthritis (PIA),35 hexadecane-induced arthritis,37 and squalen-induced arthritis36 in the rat share many common features but differ by the degree of chronic development38 (Figure 28-1). They are induced with adjuvant compounds lacking immunogenic capacity (i.e., no specific immune responses are elicited). Instead they are rapidly spread throughout the body after a single subcutaneous injection, penetrate through cell membranes into cells, and interact with yet unknown cell surface receptors and intracellular proteins. One or two weeks after injection, arthritis suddenly develops. The arthritis appears in the peripheral joints, with a similar distribution as seen in RA. Occasionally other joints are involved, but systemic manifestations in other tissues have so far not been reported.39 In certain rat strains, especially in the PIA model, the arthritis proceeds as a chronic relapsing disease. Interestingly, a systemic immune response leading to production of antibodies to RA33 and rheumatoid factors occurs, whereas no consistent immune response to specific cartilage components or citrullinated proteins has yet been observed.40,41
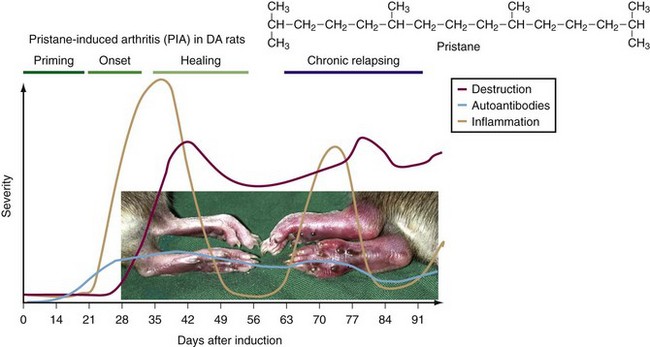
A role for cartilage proteins in regulating disease activity is, however, still possible because the disease can be prevented and in fact therapeutically ameliorated by nasal vaccination with various cartilage proteins.42 Both the initiation and the chronic progression of the arthritis is T cell dependent as shown by in vivo administration of antibodies to α/β T cells.35,36,43 Together with the observation that the chronic disease course is associated with the MHC region,35,36,43 this could implicate an activation of T cells recognizing joint-derived proteins. However, T cell transfer of the disease seems to be oligoclonal rather than monoclonal and has so far failed to identify antigen-specific T cells.38,44,45 A role for environmental infectious agents is not likely because no difference in disease susceptibility could be seen in germ-free rats, although only conventional rats respond to heat shock proteins.46 To date there is no evidence for recognition by lymphocyte receptors or receptors involved in the innate immune system. Surprisingly, some of the arthritogenic adjuvants are in fact components already present in the body before injection. For example, pristane is a component of chlorophyll and is normally ingested by all mammals including laboratory rats. Pristane is taken up through the intestine and spread throughout the body. However, they all share the capacity to penetrate into cells where they could change membrane fluidity and modulate transcriptional regulation and in higher doses induce apoptosis. The injection route and dose are critical (i.e., they determine which cell is first activated and to what extent).
The PIA model has been subjected to genetic analysis showing that the disease is polygenically controlled by at least 20 quantitative trait loci (QTL).47,48 These QTLs are often shared between the various forms of adjuvant arthritis and to a lesser degree also with CIA.49 Interestingly, they seem to control distinct phases of the disease such as arthritis onset, clinical severity, joint erosion, and chronicity.47 An approach to understand the complexity of the adjuvant arthritis, as well as the arthritis process in general, will be to eventually elucidate the underlying genetic polymorphism of these QTLs. This is, however, labor intensive and only a few genes and gene clusters have been positioned. The strongest effect is mediated through a polymorphism of the Ncf1 gene spread in both inbred strains and wild rat populations. The Ncf1 gene codes for the p47phox gene and controls the oxidative burst.50 Surprisingly, a higher oxidative burst capacity was associated with more severe arthritis. The effect was found to operate before T cell activation and therefore also controls the degree of autoimmunity, linking innate and adaptive immunity. Importantly, the MHC region, which controls the adaptive T cell response,35 and a C-type lectin gene cluster (APLEC),51 which is likely important in uptake of antigen to antigen-presenting cells, have also been identified to control PIA.
Adjuvant arthritis is not easily inducible in species other than rats. Of the previously mentioned adjuvant-induced arthritis models, only PIA has been described in the mouse.52,53 However, the induction of PIA in mouse requires repeated intraperitoneal injections of pristane, which triggers a widespread inflammatory disease with a late and insidious onset. In fact, the induced disease mimics systemic lupus erythematosus (SLE) rather than RA.54 The disease is clearly different from PIA in the rat; the same inducing protocol does not induce disease in the rat, and the disease course and characteristics are different. Another adjuvant-related model is the induction of mild arthritis after intra-articular injection of agents activating macrophages such as unmethylated DNA.26 However, it has more recently been found that several mouse models earlier believed to be spontaneous are critically dependent on adjuvants and should therefore be classified as adjuvant-induced arthritis. From the observation that a BALB/c substrain in Japan spontaneously developed arthritis, a mutation in the ZAP70 was identified.55 The ZAP70 mutation (W163C) caused weaker TCR-mediated signaling, and the development of arthritis was preceded by increased levels of IL-17–producing autoreactive T cells. The ZAP70 mutation led to defective positive selection and the emergence of autoreactive T cells attacking the joints. As a result, both rheumatoid factors and CII reactive antibodies were detected in the mice. However, the arthritis did not develop in specific pathogen free–housing conditions and it could be shown that injection of β-glycan or mannan induced the arthritis.56,57 Thus this model seems to be an adjuvant-induced arthritis in the mouse.
Other spontaneous arthritis models, the KxB/N model and the IL-1R deficient mouse, have been shown to be mediated by a likely adjuvant component because arthritis did not develop or was dramatically attenuated under germ-free conditions.58,59 The causative arthritogenic effect could be shown to be mediated by intestinal bacteria, segmented filamentous bacteria (SFB), and lactobacillus, respectively.
Taken together, there is today a set of useful adjuvant-type arthritis models in both mice and rats.
Cartilage Protein–Induced Arthritis
Arthritis is inducible with several different cartilage proteins such as aggrecan,60 link protein,61 type XI collagen CXI,62 cartilage oligomeric matrix protein (COMP),63 and CII. These various models have different characteristics and genetics. CIA, induced with CII, is today the most commonly used model for RA. It was first demonstrated in the rat64 and was later reported using other species such as mouse65 and primates.66 Today CIA is the most commonly used model for RA.
Collagen (II)-Induced Arthritis
Immunization with the major collagen in cartilage, type II collagen (CII), leads to an autoimmune response and as a consequence, sudden onset of severe arthritis. Although it is usually necessary to emulsify the CII in adjuvant such as mineral oil in the rat and complete Freund’s adjuvant in the mouse, the disease can be distinguished from the various forms of adjuvant arthritis.67 However, the CIA model varies considerably depending on the experimental animal strain, the adjuvant used, and whether CII used is of self or nonself origin.
In both rats and mice immunized with heterologous CII, a severe, erosive polyarthritis develops 2 to 3 weeks after immunization but usually subsides within 3 to 4 weeks (Figure 28-2). The most commonly used DBA/1 strain thus develops a severe but only an acute disease. However, a genetic influence is obvious because mice on C57Bl/10 backgrounds develop a milder arthritis that later may develop into a more chronic relapsing disease course68–70 (Figure 28-3). In all of the models the erosive inflammatory phase is followed by a healing phase with pronounced formation of new cartilage and bone that clinically can be difficult to distinguish from active inflammation. The disease is critically dependent on both T cell and B cell responses to CII, and pathogenic antibodies play a role in the inflammatory attack on the joints.71,72 The CAIA is inducible with certain CII-specific monoclonal antibodies73 (Figure 28-4). These CII-specific antibodies bind to the cartilage and destabilize the cartilage matrix. Subsequently the inflammatory response is triggered with infiltration of antibodies into cartilage matrix, fixation of complement attraction of neutrophilic granulocytes, and activation of FcR expressing inflammatory cells in a process independent of the immune system.74,75 Interestingly, the epitopes recognized on CII contain arginines that potentially can be citrullinated. Recently it could be shown that one of the major CII epitopes is citrullinated, and monoclonal antibodies specific for the citrullinated peptide induce arthritis showing an important link to RA.76
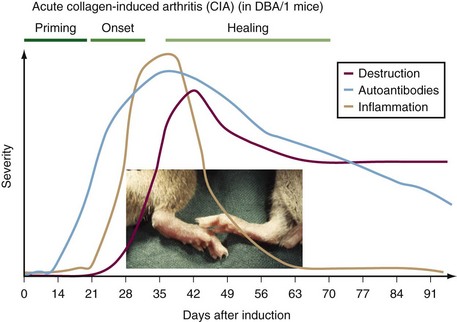
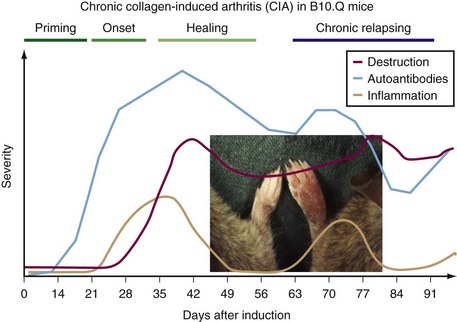
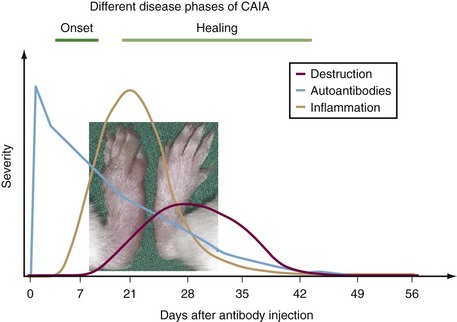
The disease induced after immunization with homologous CII in both rats and mice is not as easily inducible, but once started it is severe and tends to be more chronic than the disease induced with heterologous CII.77 The pathogenic events in the chronic disease phase are largely unknown but are most likely dependent on both autoreactive B cell and T cell activity. Nevertheless, the CIA model is the most extensively investigated model for RA and has given valuable insights into the genetic control of the arthritic process and of the autoimmune interactions with cartilage. It has also been useful for the development of new therapeutic approaches and for drug screening.
Genetic Basis of Collagen-Induced Arthritis
Susceptibility to CIA varies dramatically between different inbred strains. The CIA is a complex, polygenic disease, similar to the adjuvant arthritis models described earlier. In the CIA model the autoimmune process is already determined by the induction through immunization with a defined antigen. Not surprisingly, the MHC class II polymorphism is important for determining susceptibility,78,79 but there is also a major influence by a large number of genes outside the MHC region. The major gene regions have been identified through genetic segregation experiments in both mice and rats, which have given an overall picture of the genetic inheritance of the susceptibility.80–82 As in other complex diseases, these genes operate in concert and can only be identified through isolation in a controlled genetic and environmental context.83,84 So far MHC class II genes (Aq), Ncf1, and complement C5 have been positioned from genetic analysis. The Ncf1 gene was defined through analysis of PIA and CIA in rats.50 A spontaneous mutation in the mouse Ncf1 gene, when combined with the CIA-susceptible MHC class II allele Aq in the C57Bl/10 mouse, develops a chronic relapsing form of CIA.70 In addition, these mice tend to develop a severe form of chronic arthritis in the postpartum period, with the spontaneous development of autoimmunity to CII.70 Another genetic polymorphism of importance is the complement C5, which is deficient in many mouse strains. The deficiency leads to a relative resistance to CIA, suggesting a role for complement pathways in arthritis,85 which in fact is opposite of its role in mycoplasma-induced arthritis.86 A role for alternative complement pathways and Fc receptor–mediated pathways has been demonstrated using both CIA and the CAIA models.87–91 The rapid progress of genome-wide association analysis of large human cohorts today gives direct information on involved genes and gene clusters in common diseases such as RA. However, the animal models are necessary to understand their functional relevance; for this, the genes controlling the corresponding disease in the animals need to be identified.92 Knowledge of animal model genetics will facilitate creation of humanized models through genetic modification.
Role of the Major Histocompatibility Complex
Early observations using the CIA model in both mice and rats indicated a role for the MHC region. In the mouse, CIA induced with either heterologous or homologous CII is most strongly associated with the H2q and H2r haplotypes, whereas most other haplotypes such as b, s, d, and p are relatively resistant.79 The major underlying gene within the H2q haplotype has been identified as Aq beta.78 Moreover, the immunodominant peptide derived from the CII molecule bound to the arthritis-associated q variant of the A (Aq) molecule has been found to be located between positions 259 and 271 of CII.93,94 This peptide can be glycosylated on the central lysine side chain and is recognized by most of the CII-reactive T cells.95 Interestingly, the peptide is also bound by DR4 (DRB1*0401/DRA) and DR1 molecules (i.e., the shared epitope), which are associated with RA. Mice transgenically expressing DR4 or DR1 are susceptible to CIA and respond to CII259-271 peptide,96,97 and CII-reactive T cells from RA patients seem to predominantly recognize the glycosylated forms of the CII259-271 peptide.98 These findings suggest a model for studies of RA that not only mimic some basic pathogenic events but may also share some critical structural similarities.
Arthritis is also inducible in mouse strains that do not express q or r. A commonly used model is to induce arthritis with high doses of chicken or bovine CII emulsified in Mycobacterium tuberculosis–containing CFA.99 T cell autoreactivity to CII has not yet been reproducibly demonstrated, however, and it is possible that the T cell reactivity is directed to a contaminant in the preparation such as another matrix protein or the pepsin used for the preparation. Such T cell reactivity could help B cells produce antibodies to CII, explaining the development of arthritis.
Autoimmunity in Collagen-Induced Arthritis
It is important to emphasize that the identified structural interaction between MHC class II–positive peptide complexes and T cells does not give us the answer to the pathogenesis of CIA (or RA), but rather a better tool for further analysis. An important question is how the immune system interacts with the peripheral joints (i.e., how autoreactive T and B cells are normally tolerized and what happens in the pathologic situation, after their activation by CII immunization). Most of the T cells reactive with the rat CII259-271 peptide do not cross-react with the corresponding peptide from mouse CII. The difference between the heterologous and the homologous peptide is position 266 in which the rat has a glutamic acid (E) and the mouse an aspartic acid (D), which leads to a weaker binding of the mouse peptide to Aq. The importance of this minor difference was demonstrated in transgenic mice expressing CII mutated to express a glutamic acid at this position.100 When mutated CII was expressed in cartilage, the T cell response to CII was partially but not completely tolerized. The mice were susceptible to arthritis, but the incidence was low, similar to what is seen in mice immunized with homologous CII. This finding shows that a normal interaction between cartilage and T cells leads to the activation of T cells, but with less capacity to induce arthritis or with regulatory properties. These CII autoreactive T cells may under extreme circumstances (such as CII immunization) be pathogenic. In contrast, B cells reactive with CII are not tolerized and as soon as the T cells are activated, even in a partially tolerized state, they may help B cells to produce autoreactive and pathogenic antibodies. It is possible that a similar situation in humans could explain the difficulties in isolating CII-reactive T cells compared with the relative ease in which CII-reactive B cells can be detected in the joints.
Induction of Arthritis with Other Cartilage and Joint-Related Proteins
Type XI Collagen-Induced Arthritis
The type XI collagen (CXI) is structurally similar to CII and is to a large extent co-localized. CXI is a heterotrimer with three different α chains, one of which is shared with CII (the α3 chain). Both heterologous and homologous CXI have been reported to induce arthritis in rat strains.101,102 Interestingly, the induction with homologous CXI gives a chronic relapsing disease, which is distinctly different from the heterologous CXI-induced disease and CII-induced CIA.
COMP-Induced Arthritis
Another cartilage protein is cartilage oligomeric matrix protein (COMP). Homologous COMP induces arthritis in both rats and mice.63,103 In comparison with CIA, the resulting disease is self-limited, is less erosive, and has a different genetic control.
Proteoglycan (Aggrecan)-Induced Arthritis
Other major components of joint cartilage are proteoglycans, of which the largest is aggrecan. Immunization of Balb/c mice with fetal human aggrecan induces chronic arthritis.60 Both B and T cells are involved in the pathogenesis. Autoreactive T cells have been isolated and respond to the G1-domain of aggrecan in which neoepitopes are created104 and T cell receptor transgenic mice spontaneously develop arthritis at high age.105 The disease has been genetically mapped and shown to share many gene regions in common with CIA.106

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


