119 Hemophilic Arthropathy
Severe hemophilia, if not aggressively treated, is often complicated by recurrent hemarthrosis.
Soft tissue and muscle hemorrhage are frequent complications of hemophilia.
Although spontaneous joint hemorrhage has been described in a variety of inherited disorders of coagulation1–3 and in the setting of anticoagulation therapy,4 it occurs most frequently in hemophilia. Bleeding into the joints is the complication of hemophilia that most often requires therapeutic intervention and, when it is recurrent, can lead to chronic, deforming arthritis that is independent of bleeding episodes.
Although the intrinsic pathway of coagulation is severely impaired in hemophilia, the extrinsic tissue–dependent pathway remains intact and is probably the major hemostatic regulatory system. Normal synovial tissue and cultures of synovial fibroblasts have been found to be deficient in tissue factor,5 which suggests that in synovium-lined joints, hemophiliacs have functional inactivity of intrinsic and extrinsic coagulation pathways. This observation may explain the marked propensity toward hemorrhage in joints compared with other tissue sites in these patients.
Clinical Features
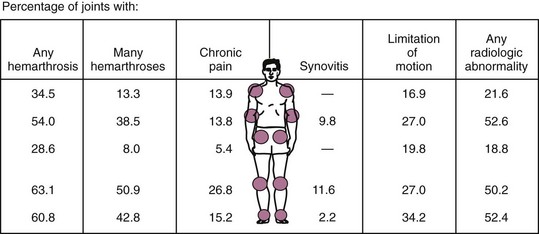
The spectrum of articular disease in hemophilic patients has been the subject of numerous comprehensive reviews6–10 and includes acute hemarthrosis, subacute or chronic arthritis, and end-stage hemophilic arthropathy. The usual distribution of joint involvement is shown in Figure 119-1. Involvement of the small joints of the hands and feet also may occur, although infrequently.
Septic Arthritis
Until the early 1980s, septic arthritis rarely occurred in hemophilic patients. With widespread occurrence of human immunodeficiency virus (HIV) infection as a result of contaminated factor concentrates, the incidence of this complication has increased significantly.11,12 Septic arthritis is seen more often in adult than in pediatric hemophilic patients and is most commonly monoarticular, usually involving the knee. In contrast to spontaneous hemarthrosis, septic arthritis is significantly associated with a temperature greater than 38° C within 12 hours of presentation and articular pain that does not improve with replacement therapy.11 Peripheral leukocyte count may not be elevated, particularly in HIV-positive patients.13 A predisposing factor other than hemophilic arthropathy is often identifiable, including previous arthrocentesis or arthroplasty, intravenous drug use, and an infected indwelling venous access catheter. Staphylococcus aureus is the most frequently identified organism even in HIV-infected patients, followed by Streptococcus pneumoniae.13
Muscle and Soft Tissue Hemorrhage
Bleeding into muscles and soft tissue is common in hemophilic patients and may be more insidious than hemarthrosis because of the lack of premonitory symptoms. Bleeding into the iliopsoas and gastrocnemius muscles and the forearm results in well-described syndromes with which the rheumatologist should be familiar. Iliopsoas hemorrhage produces acute groin pain with marked pain on hip extension and a hip flexion contracture. Rotation is preserved, in contrast to intra-articular hemorrhage. If untreated, the expanding soft tissue mass may compress the femoral nerve, causing signs and symptoms of femoral neuropathy.6,14 Bleeding into the gastrocnemius muscle can cause an equinus deformity from heel cord contracture.6 Finally, hemorrhage into closed compartments can cause acute muscle necrosis and nerve compression.15 Of particular importance is bleeding into the volar compartment of the forearm, which can cause flexion deformities of the wrist and fingers. If a compartment syndrome is suspected, compartment pressures should be measured to confirm the diagnosis.
A large intramuscular hemorrhage uncommonly results in the formation of a simple muscle cyst, which clinically appears to be an encapsulated soft tissue area of swelling overlying muscle. Cyst formation in this setting is confined by the muscular fascial plane and most likely results from inadequate resorption of blood and clot. Subperiosteal or intraosseous hemorrhage, in contrast, may lead to a pseudotumor, a rare skeletal complication of hemophilia. Hemophilic pseudotumors are of two types: the adult type, which occurs proximally, usually in the pelvis or femur; and the childhood type, which occurs distal to the elbows or knees and carries a better prognosis.16,17
Conservative early management of muscle cysts and childhood-type pseudotumors is indicated, including immobilization and factor replacement. In adult-type pseudotumors, which usually are refractory to conservative therapy, and in progressive childhood pseudotumors, surgical removal is indicated16 to prevent serious complications, such as spontaneous rupture, fistula formation, neurologic or vascular entrapment, and fracture of adjacent bone. Aspiration of a pseudotumor or cyst is contraindicated.
Diagnostic Imaging
Radiographs
With progression of chronic proliferative synovitis, irreversible radiologic changes appear.18 These changes reflect the inflammatory and degenerative nature of chronic hemophilic arthropathy (Table 119-1 and Figure 119-2A). Certain changes unique to hemophilic arthropathy occur as well (see Table 119-1 and Figure 119-2B). Radiographic findings in hemophilic arthropathy have been recently reviewed.18a
Table 119-1 Radiologic Manifestations of Chronic Hemophilic Arthropathy
| Characteristic | Also Seen in |
|---|---|
| Periarticular soft tissue swelling | RA |
| Periarticular demineralization | RA |
| Marginal erosions | RA |
| Subchondral irregularity and cyst formation | RA, OA |
| Decreased joint space | OA |
| Osteophyte formation | OA* |
| Chondrocalcinosis | CPPD |
| Specific |
CPPD, calcium pyrophosphate deposition disease; OA, osteoarthritis; RA, rheumatoid arthritis.
* From Jensen PS, Putnam CE: Chondrocalcinosis and hemophilia, Clin Radiol 28:401, 1977.
† From Schreiber RR: Musculoskeletal system: radiologic findings. In Brinkhous KM, Hemker HC, editors: Handbook of hemophilia I, New York, 1975, Elsevier.
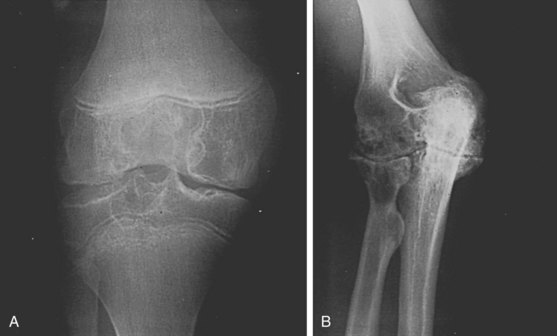
A study of serial radiographs of symptomatic joints in hemophilic patients suggests that serial scoring with conventionally accepted techniques may be a cost-effective alternative to magnetic resonance imaging (MRI) in predicting progressive synovial hypertrophy.19
Other Imaging Methods
MRI is now routinely used to stage hemophilic arthritis accurately to determine optimal treatment and to follow response to therapy.20 A scoring system based on MRI has been proposed.21 MRI has been demonstrated to be superior to conventional radiography and computed tomography in the detection of erosions and cysts, bone marrow edema and hemorrhage, and synovial hypertrophy and torn ligaments (the latter in patients whose radiographs showed widened intercondylar notches).22 As preventive measures improve, it will continue to be important to use the most advanced techniques, including MRI, both to stage hemophilic arthropathy and to assess the benefits of new therapies such as prophylactic infusion.23 Additionally, MRI and ultrasonography are useful in the detection and quantitation of soft tissue bleeding, cysts, and pseudotumors.24,25
Pathologic Features and Pathogenesis
Pathologic studies of human hemophilic arthropathy have been limited to synovial specimens obtained at surgery26,27 or at postmortem examination and reflect changes of advanced disease only. Studies of experimentally produced hemarthrosis in animals,28,29 post-traumatic hemarthrosis in nonhemophilic humans,30 and canine and murine models of hemophilia A31–33 have provided an understanding of the earliest changes induced by acute hemarthrosis and their evolution to chronic arthritis.
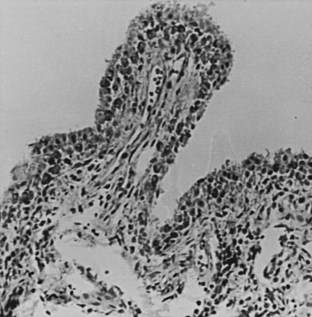
As reviewed more recently,34 the process most likely includes catabolic activation of synovial cells by exposure to blood components with subsequent cartilage destruction and a direct destructive effect of intra-articular blood on cartilage. A single synovial hemorrhage induces serial changes in the synovial membrane, including early focal villous synovial proliferation and subsynovial diapedesis of erythrocytes, followed by the appearance of perivascular inflammatory cells, patchy subsynovial fibrosis, and intracellular iron accumulation in synovial cells and subsynovial macrophages. With repeated hemarthroses, the synovium becomes grossly hypertrophied and hyperpigmented, with eventual organization into a pannus that invades and erodes marginal cartilage. On histologic examination, villous hypertrophy and subsynovial fibrosis progress, but inflammatory cells are scarce (Figure 119-3).27 Seventy-five percent of synoviocytes contain siderosomes (electron-dense, iron-filled deposits within lysosomes), in contrast to 10% in normal synovium and 25% in rheumatoid synovium.35 Iron deposits are associated with the production of proinflammatory cytokines and synovial inhibition of the formation of human cartilage matrix. Although the inflammatory synovial changes are mild, the synovial production of proinflammatory mediators, including interleukin-1 and interleukin-6 and tumor necrosis factor, approaches that of rheumatoid synovium.34 The articular cartilage is grossly and microscopically abnormal in the setting of recurrent hemarthrosis.28 Areas of cartilaginous fissuring and rarefaction expose sclerotic bone. The remaining cartilage is thin and unevenly distributed, often freely protruding into the joint cavity. Bone erosions appear at weight-bearing surfaces. Loss of matrix glycosaminoglycan occurs and is also seen in degenerative arthritis.27,36
Current studies suggest that recurrent hemarthrosis induces joint destruction in hemophilic arthropathy through direct and indirect effects of iron on the synovium35,37 and cartilage,36,38 by the degradative effect of proliferative synovium,39 and through an alteration in cartilage biochemical composition similar to that seen in degenerative arthritis. A relationship between hemarthrosis-induced overexpression of oncogenes (e.g., c-myc and mdm-2) and the dysregulated, tumor-like proliferation of hemophilic synovium has been noted.40–42 Angiogenesis induced by growth factors such as vascular-derived endothelial growth factor may be important in inducing and sustaining a synovial proliferative response to hemarthrosis.42
< div class='tao-gold-member'>




