107 Treatment of Juvenile Idiopathic Arthritis
Juvenile idiopathic arthritis (JIA) is the most common rheumatic disease in childhood, but actual estimates of prevalence and incidence vary remarkably in different geographic regions, ranging from 7 to 400 per 100,000 children, reflecting variations in disease reporting, classification, and ethnic and environmental differences in disease expression.1 Reasonable working estimates are 150 per 100,000 children, which makes JIA one of the most common chronic diseases of childhood. There are significant differences in the disease manifestations in children compared with adults, with some types occurring exclusively in children. This chapter discusses the current understanding of the key clinical features of the various forms of JIA, differential diagnoses, treatment approaches, prognosis, and outcomes. The rapid growth in understanding the biologic basis for JIA and the ongoing development of targeted therapies for rheumatic diseases are likely to lead to enhancements in these recommendations. Data concerning long-term disease course and outcome in children with JIA in this modern treatment era remain limited, and there are international prospective inception cohorts of children with arthritis designed to address these gaps in knowledge.2,3
Classification Criteria for Juvenile Idiopathic Arthritis
Differential Diagnosis
In the past, different groups had used various types of nomenclature to classify children with persistent arthritis including “juvenile rheumatoid arthritis” (American College of Rheumatology [ACR]) and “juvenile chronic arthritis” (European League Against Rheumatism), which created problems in comparing research studies and outcomes. The goal of the International League of Associations for Rheumatology (ILAR) is to identify subtypes of JIA for research purposes that are homogeneous and mutually exclusive. JIA classification is currently based on predominant clinical and laboratory features and the number of involved joints at disease onset.4 There is a continual renewal process, with the second revision occurring in Edmonton in 2001, which is presented in Table 107-1. However, classification systems are ever evolving, and categorization may evolve to more biologically and genetically similar subgrouping, especially with recent advances in etiology and pathogenesis. For example, age of onset may be a more biologically relevant parameter to distinguish between subtypes of JIA than classification based on number of involved joints. PBMC gene expression analysis reveals biologic differences between patients with early-onset (<6 years) and late-onset (>6 years) JIA, which was independent of oligoarthritis or polyarthritis subtype.5 Ravelli and colleagues6 provided clinical support for this approach, showing that antinuclear antibody (ANA)-positive patients with oligoarthritis and rheumatoid factor (RF)-negative polyarthritis were similar in terms of early age at onset, female predilection, increased frequency of asymmetric arthritis, and increased frequency of uveitis.
Table 107-1 International League of Associations for Rheumatology Classification Criteria for Juvenile Idiopathic Arthritis (JIA)4
| General definition of JIA: arthritis of unknown etiology that begins before the sixteenth birthday and persists for at least 6 wk; other known conditions are excluded |
| Subcategory | Definition | Exclusions |
|---|---|---|
| Oligoarthritis | Arthritis affecting 1-4 joints during the first 6 mo of disease | a. Psoriasis or a history of psoriasis in the patient or first-degree relative b. Arthritis in an HLA-B27–positive male beginning after the sixth birthday c. Ankylosing spondylitis, ERA, sacroiliitis with inflammatory bowel disease, reactive arthritis, acute anterior uveitis, or a history of one of these disorders in a first-degree relative d. The presence of IgM RF on at least 2 occasions at least 3 mo apart |
| RF-Negative Polyarthritis | a, b, c, d, e | |
| RF-Positive Polyarthritis | a, b, c, e | |
| Psoriatic Arthritis | b, c, d, e | |
| Enthesitis-Related Arthritis | a, d, e | |
| Systemic JIA | Arthritis in 1 or more joints with, or preceded by, fever of at least 2 weeks’ duration that is documented to be daily and quotidian (fever that rises to ≥39° C once a day and returns to ≤37° C between fever peaks) for at least 3 days, and accompanied by 1 or more of the following: | a, b, c, d |
| Undifferentiated Arthritis | Arthritis that fulfills criteria in no category or in ≥2 of the above categories |
ERA, enthesitis-related arthritis; RF, rheumatoid factor.
Among patients with acute lymphocytic leukemia (ALL), 15% to 30% present with musculoskeletal symptoms and may be misdiagnosed as JIA.7 Up to 75% of children ultimately diagnosed with ALL presenting with musculoskeletal complaints did not have blasts in the peripheral blood at the time of evaluation by pediatric rheumatologists, although low white blood cell count, mild thrombocytopenia, and nighttime pain were early indicators of ALL. ANA status, rash, radiographic abnormalities, and objective signs of arthritis were not helpful in distinguishing between ALL and JIA because they occurred at similar rates in both groups.8
Each subcategory is discussed later, focusing on clinical manifestations, diagnostic features, treatment, outcome, and prognosis. Treatment recommendations are discussed later for each of the subtypes of JIA on the basis of current available evidence including the recently developed ACR recommendations for the treatment of JIA.9
Rheumatoid Factor–Negative Polyarthritis
Clinical Manifestations and Diagnostic Features
RF-negative polyarthritis makes up 10% to 30% of all JIA cases, with a bimodal distribution of age of onset with the first peak at 1 to 4 years of age and the second peak at 10 to 12 years. Girls are more commonly affected than boys with a ratio of 3.2 : 1, and subacute anterior uveitis occurs in 4% to 25%.10 Any joint may be affected in RF-negative polyarthritis JIA, with more involvement of hip, shoulder, cervical spine, and distal interphalangeal joints than in adults. The arthritis is often usually insidious and can be symmetric or asymmetric, affecting both large and small joints. Some authors distinguish between two clinical subgroups on the basis of ANA status: (1) an ANA-positive form that resembles oligoarthritis, except for the number of joints affected in the first 6 months of disease, consisting of young girls (younger than age 6) with an asymmetric-onset arthritis and at a high risk of uveitis, and (2) an ANA-negative form that is similar to adult-onset RF-negative rheumatoid arthritis (RA), characterized by symmetric synovitis of large and small joints, with onset in a slightly older age group (ages 7 to 9). The similarities between ANA-positive, RF-negative polyarthritis and oligoarthritis have led to the hypothesis that these two entities are actually in the same disease spectrum.6
RF-negative polyarthritis may be associated with elevated acute phase reactants, mild anemia, and ANA positivity in up to 40%. Even though RF is negative, 50% to 80% of patients are ACPA positive,10 using high-sensitivity (but low-specificity) testing methods.11
Treatment
With the current development of increasingly more effective biologic treatment for arthritis, pediatric rheumatologists now aim to achieve complete disease remission as early as possible in the disease course. Several studies support the paradigm of treating aggressively to reach inactive disease as early as possible, which may ultimately lead to better outcome such as improved quality of life, shorter periods of time spent in active disease, and less long-term joint damage. Active disease in the first 2 years was significantly associated with the duration of active disease in the following 3 years,12 and conversely, improved disease control with disease-modifying antirheumatic drugs (DMARDs) and/or biologics was associated with improved outcomes.13 To this end, children with polyarthritis require a disease-modifying agent as soon as practically possible after the diagnosis has been confirmed. If nonsteroidal anti-inflammatory drugs (NSAIDs) are initially used as monotherapy, with or without intra-articular steroids (IASs), continued disease activity at no more than 2 months should prompt escalation of therapy.9 As shown in Figure 107-1, methotrexate (MTX) is the first DMARD of choice, but if there is an inadequate response to MTX by 2 months, anti–tumor necrosis factor (TNF) agents should be initiated.14,15 The different TNF inhibitors have not been directly compared against each other, so it is not possible to determine which might be most effective and safe in a given patient. Switching between TNF inhibitors in children has not been well studied. Sulfasalazine and leflunomide can still be used before an anti-TNF agent in mild disease, although evidence suggests that leflunomide may be slightly less effective than MTX.16 Polyarticular disease course of any type, which does not respond well to MTX or anti-TNF agents, is now increasingly being treated with a range of newer biologics. Physiotherapy (PT) is important for all children with JIA, for stretching, muscle building, and consequent joint protection. Children with hand involvement need occupational therapy (OT) assessment and input regarding writing and school accommodations. The following paragraphs summarize specific information about different therapies used in children and may be applied to all types of JIA as appropriate. (See Table 107-2 for medications, dosing, route, and safety monitoring recommendations.)
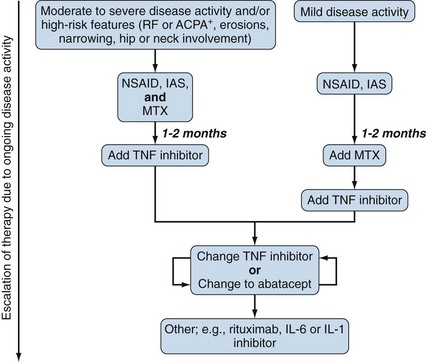
Table 107-2 Commonly Used Medications in Juvenile Idiopathic Arthritis9,176
| Medication | Typical Maximum Dose | Typical Frequency |
|---|---|---|
| Abatacept | 10 mg/kg (max 1000 mg) IV | Load at 0, 2, 4 wk, then every 4 wk |
| Adalimumab | 24 mg/m2 <30 kg: 20 mg SQ >30 kg: 40 mg SQ | Every 2 wk |
| Anakinra | 2 mg/kg (max 100 mg) SQ | Daily |
| Cyclosporine | 6 mg/kg/day orally | Divided twice a day |
| Diclofenac (SR preparation available) | 1-3 mg/kg/day (max 150 mg/day) | Divided 1-3 times a day |
| Etanercept | 0.8 mg/kg/wk (max 50 mg)* SQ 0.4 mg/kg/dose (max 25 mg) SQ | Once weekly 2×/wk |
| Ibuprofen | ≥6 mo of age: 30-40 mg/kg/day | Divided 3-4 times a day |
| Indomethacin (SR preparation available) | >1 mo of age: 1-2 mg/kg/day (max 50 mg/day) | Divided 1-2 times a day |
| Infliximab | 10 mg/kg/dose IV | Load at 0, 2, 6 wk, then every 4 wk |
| Intravenous immunoglobulin | 2 g/kg/dose IV | Every 2 wk |
| Leflunomide | <40 kg: 10 mg orally >40 kg: 20 mg orally | Daily |
| Methotrexate | 15 mg/m2/dose SQ (0.6 mg/kg; max 25 mg) | Weekly |
| Naproxen | >2 yr of age: 20-30 mg/kg/day (max 1 g/day) | Divided twice a day |
| Piroxicam | <15 kg: 5 mg orally 16-25 kg: 10 mg orally 26-45 kg: 15 mg orally >46 kg: 20 mg orally | Daily |
| Rilonacept | 2.2-4.4 mg/kg SQ | Once weekly |
| Rituximab | 750 mg/m2 (max 1000 mg) IV | Twice 2 wk apart |
| Sulfasalazine | 50 mg/kg/day (max 2 g) orally | Divided twice a day |
| Tacrolimus | 0.2 mg/kg/day orally | Divided twice a day |
| Thalidomide | 5 mg/kg/dose orally | Daily |
| Tocilizumab | <30 kg: 12 mg/kg IV >30 kg: 8 mg/kg IV | Every 2 wk |
| Summary of Recommendations for Medication Safety Monitoring | ||
| Nonsteroidal Anti-inflammatory Drugs | ||
| Methotrexate | ||
| Tumor Necrosis Factor Inhibitors | ||
IV, intravenous; SQ, subcutaneous; SR, sustained release.
* The effectiveness of the 0.8 mg/kg/wk dose has been evaluated in JIA patients.177,178
Nonsteroidal Anti-inflammatory Drug Use in Children
NSAIDs may help control symptoms but do not alter the natural history of JIA. In general, NSAIDs should only be considered as monotherapy in initial therapy in low disease activity. If control is not achieved in 1 to 2 months, additional therapy should be considered.9 NSAIDs are frequently used for symptom control as an adjunctive therapy to more definitive therapies. Gastric protection with H2 blockers or proton pump inhibitors may be required.
Intra-articular Steroid Injections
The use of intra-articular triamcinolone hexacetonide (THA), 1 mg/kg in large joints such as the knee and 0.5 mg/kg in smaller joints such as the ankle, has been found to be superior to triamcinalone acetonide, betamethasone, and methylprednisolone acetate in randomized controlled trials.17–22 Early treatment is associated with better outcome,23 and IASs are expected to result in clinical improvement of arthritis for at least 4 months. Therefore if arthritis recurs, joint injections can be repeated up to three times in a 12-month period. Difficult-to-reach joints such as the hip, sacroiliac (SI) joint, temporomandibular joint (TMJ), and subtalar joint may be injected using ultrasound or fluoroscopy. IASs should be administered under sedation or general anesthesia for the young. The use of IASs for active arthritis has been recommended by the ACR guidelines regardless of concurrent therapy, JIA subtype, disease activity, prognostic features, or joint contracture. If the duration of clinical improvement is shorter than 4 months, systemic treatment (e.g., MTX) may be indicated.9
Corticosteroid Use in Children with Juvenile Idiopathic Arthritis
In general, systemic corticosteroids should be used sparingly in the treatment of any subgroup of JIA because of the severe morbidity associated with chronic use, even at low doses. Newer therapeutic modalities such as biologics reduce the dependence for any corticosteroids and/or limit the doses needed. The evidence for use of corticosteroids for the synovitis of JIA is controversial and conflicting, and the ACR recommendations for JIA treatment were unable to make any recommendations for corticosteroid use, except in systemic juvenile idiopathic arthritis (sJIA) for severe systemic features.9 However, in some cases, low doses of corticosteroids (<0.1 mg prednisone equivalent/kg/day) or brief high-dose regimens (intravenous methylprednisolone 30 mg/kg/day for 1 to 3 days) may be used in polyarticular JIA, in order to bridge constitutional features of pain and fatigue while waiting for DMARDs or biologic therapies to reach their therapeutic effect. Local use of corticosteroids for uveitis is discussed later in “Uveitis.”
Methotrexate
MTX is the most commonly used DMARD in JIA.24 In a retrospective cohort study involving all JIA subtypes, the strongest predictor of response to MTX at 6 months of treatment was the time from diagnosis to start of MTX, suggesting that starting MTX early will lead to a better response.25 The ACR guidelines support a maximum dose of 0.6 mg/kg once weekly (equivalent to 15 mg/m2/week, maximal 25 mg/week) of parenteral MTX.9 In patients with lower disease severity, lower doses (8 to 12.5 mg/m2/week oral or parenteral) may be effective, and these doses are similar in safety.15 Most pediatric rheumatologists start folic acid at 1 mg/day, but daily folate supplementation remains controversial. Routine monitoring of liver enzyme tests should be done as noted in Table 107-2, but liver biopsies are not indicated except in unusual circumstances. Some children develop an intolerance to MTX, and leflunomide may be used as an alternative.16,26 Both MTX and leflunomide are associated with teratogenic effects.
Tumor Necrosis Factor Inhibitors
Anti-TNF agents are effective in many children with polyarticular-course JIA of any onset type who fail to respond fully to MTX. The first TNF inhibitor to be studied in JIA, etanercept, demonstrated efficacy and safety in a novel randomized withdrawal trial design in patients with polyarticular JIA, in which the study design was based on an open-label period, followed by randomization of responders to placebo or study drug with the primary endpoint being time to flare.14 Since that initial randomized control study in 2000, long-term studies and registries have continued to demonstrate the safety and efficacy of etanercept in children with polyarthritis, with and without MTX.27–32 The frequency of major serious adverse events (SAEs) have been low, with adjusted rates of 0.12 events per patient year and 0.03 medically important infections per patient year in open-label extensions.30 Importantly, no cases of lupus, demyelinating disorders, malignancies, opportunistic infections, or tuberculosis were seen. Although there have been several long-term studies of etanercept with international registries, the total number of patient-years available for evaluation is still too small to be able to detect rare long-term SAEs such as malignancy or demyelinating diseases.
A 1-year prospective, observational study to compare combination etanercept and MTX with etanercept monotherapy demonstrated that the likelihood of achieving 70% disease control (ACR Pedi 70) was increased with combination therapy (odds ratio of 2.1, 95% CI 1.2 to 3.5) as compared with etanercept monotherapy.27 In this cohort, there were 24 infectious SAEs and 23 noninfectious SAEs including three malignancies in 496 patients over 12 months. Growth delay in JIA was improved and reversed with the use of etanercept.33
An international, multicenter, randomized, double-blind, placebo-controlled trial of infliximab provided important lessons regarding dosing in children.34 The initial dose of 3 mg/kg/infusion based on adult studies did not demonstrate efficacy at 3 months compared with placebo; however, a separate arm suggested that 6 mg/kg/infusion had better pharmacokinetics leading to better effectiveness. In a 4-year, long-term, open-label extension, 14% of patients discontinued infliximab due to SAEs including six infectious SAEs in 120 patients over 52 weeks.35 Significant infusion reactions associated with infliximab antibodies and asymptomatic development of antinuclear antibodies were observed.
Adalimumab was shown to be efficacious and safe in a randomized placebo-controlled withdrawal trial in patients with moderately to severely active polyarticular JIA with or without MTX, and there was a trend toward more improvement with combination therapy, although the study was not statistically powered to measure this difference.36 The dose used was adalimumab 24 mg/m2 (maximum dose, 40 mg) subcutaneously every other week. SAEs possibly related to adalimumab occurred in 14 patients (total 177), seven of which were serious infections.
In August 2009, the U.S. Food and Drug Administration issued a black box warning on the increased risk of cancer, particularly lymphoma, in children and adolescents receiving TNF inhibitors for arthritis or inflammatory bowel disease (IBD).37 The other adverse events involved with TNF inhibitors are similar to that of adults and have been reported in children as well including serious infections, demyelinating processes, optic neuritis, injection site reactions or infusion reactions, and development of autoimmune conditions. Because of the voluntary nature of reporting rare adverse drug events in the United States, the actual risk of any of these rare events is not known.
Abatacept
The T cell co-stimulatory inhibitor, CTLA4-Ig (abatacept), was studied in an international placebo-controlled randomized withdrawal trial in 190 patients with active polyarthritis regardless of onset type who had inadequate response or intolerance to one DMARD in the past including TNF inhibitors.38 Concurrent MTX was allowed. Thirty-percent improvement (ACR Pedi 30) response rates were 76% in biologic-naïve patients and 39% in patients with prior biologic therapy. Abatacept continued to be clinically significant with durable efficacy in patients with JIA, and some patients require longer periods for optimal response (>3 to 4 months) compared with TNF inhibitors.39 No cases of tuberculosis or malignancy were detected, but patient numbers were small and follow-up time was limited.
Other Biologics
The ACR guidelines recommend consideration of rituximab as a treatment option for patients who have received TNF inhibitor and abatacept sequentially and have high disease activity.9 IL-6 and IL-1 inhibition are discussed in more detail later in “Systemic Juvenile Idiopathic Arthritis” but have not been specifically studied in polyarticular disease. The use of nonbiologic DMARD combinations (such as MTX plus sulfasalazine and/or hydroxychloroquine) has not been studied in children.
Outcome
Early response to treatment was an important predictor of long-term outcome.13 Symmetric arthritis and early hand involvement predicted future disability and poorer overall well-being.40 The ACR guidelines for JIA treatment also consider the following as poor prognostic factors in patients with RF-negative polyarthritis: arthritis of hip or cervical spine; positive ACPAs; and radiographic damage (erosions or joint space narrowing by radiograph).9
RF-negative polyarthritis has a variable outcome, which shows the heterogeneity of the subtype,41 but the overall prognosis appears to be better than RF-positive polyarticular JIA.42 Approximately 30% of children will go into long-term remission off medication, with the chance of remission being highest in the first 5 years of disease.43,44 However, flare of disease 2 years after reaching clinical remission off medication had occurred in 69% of patients.43
Rheumatoid Factor–Positive Polyarthritis
Clinical Manifestations and Diagnostic Features
RF-positive polyarthritis is a well-characterized JIA subcategory and is part of the same disease spectrum as adult RF-positive RA,45 sharing immunogenetic and serologic factors. This subcategory makes up 5% to 10% of cases of JIA10 and is more common in girls, with reported female-to-male ratios between 5.7 and 12.8 : 1.0.46,47 Age of onset occurs in late childhood or adolescence and typically is an aggressive, symmetric polyarthritis affecting the small joints of the hands, as well as large joint involvement in a pattern that resembles RA. These children frequently have more than 30 joints with arthritis. Hip involvement is common and may be debilitating. The arthritis can be quite severe, often resulting in bony erosions and joint destruction. Radiologic changes tend to take place early,42 especially in hands and feet. With active disease, patients may occasionally have mild systemic signs and symptoms such as weight loss, low-grade fever, malaise, mild hepatosplenomegaly, or lymphadenopathy. Rheumatoid nodules occur in up to 10% of cases, most frequently around the elbow. Other extra-articular manifestations are reported less often than in adults. Uveitis is an unusual feature of this subtype, occurring in only about 0% to 2% of patients.10
Polyarthritis may be associated with mild to moderate inflammation such as elevated acute-phase reactants and a normocytic, normochromic anemia. By definition, all patients have IgM-anti-IgG RF. The ANA test is positive in about 55% of patients,10 and ACPAs have been reported in 57% to 73%.48,49
Treatment
Treatment algorithms and ACR guidelines for patients with all types of polyarthritis are discussed in Figure 107-1. However, because children with RF-positive polyarthritis are at higher risk of prolonged erosive arthritis compared with other types of JIA, these children should be considered to be in the more severe disease category, requiring rapid escalation of treatment if even mild disease activity persists. Rather than an initial period of NSAID monotherapy, RF-positive polyarthritis patients should receive MTX, at the time of diagnosis, with rapid addition of a TNF inhibitor if response is not adequate.9 Some children benefit from multiple joint injections to maintain control of the arthritis.
Outcome
Children with RF-positive polyarthritis have a poorer long-term prognosis than the other JIA subcategories.42,50 Inactive disease is difficult to achieve with 84% of the disease course consisting of active disease, and only 5% of patients were able to maintain remission after cessation of therapy.43
Oligoarticular Juvenile Idiopathic Arthritis
Clinical and Diagnostic Features
Oligoarticular JIA accounts for up to 20% of all new rheumatic diagnoses in the general pediatric rheumatology clinic47 and is the most prevalent of all the JIA subcategories, comprising 30% to 60% of all JIA patients in North America and Europe.41 Oligoarticular JIA has no adult equivalent. The peak age of onset occurs in Caucasian children ages 2 to 4 years from the United States and Europe.10 Females are affected more commonly than males, 3 : 1.10 Two general subgroups are recognized within oligoarticular JIA: extended oligoarticular involvement, in which many additional joints develop arthritis after the initial 6 months, as contrasted to persistent oligoarticular JIA, in which the number of joints affected remains less than 5. Currently, there is no single reliable predictor of extension, but symmetric disease, ankle and/or wrist involvement, and an elevated erythrocyte sedimentation rate (ESR) in the first 6 months of disease may indicate likelihood of extension.51,52 Disease extension to the extended subtype has been reported to be 30% to 50% at 4 to 6 years after disease onset.51,53
Oligoarticular JIA usually presents as an asymmetric arthritis affecting one or two large joints, especially of the lower extremities, with the knee being the most commonly affected, followed by the ankle, wrist, and digits. Hand involvement is the third most commonly affected location, but this pattern may portend the later onset of psoriatic arthritis.54 Involvement of the hip and back, especially in young children, is so unusual that extensive evaluation is warranted to rule out other conditions such as infection or tumors.
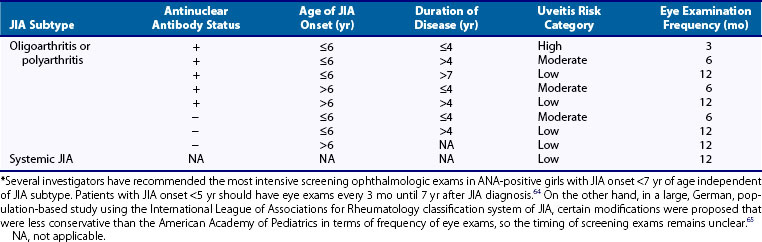
Significant constitutional and systemic symptoms are unusual in oligoarticular JIA and, if present, should raise concern regarding the accuracy of the initial diagnosis. Pain in an obviously inflamed joint is surprisingly minimal compared with septic arthritis, and in up to 25% of cases the symptoms may be subtle with parents only noticing a limp and joint swelling. In a young child there is reluctance to walk and bear weight with a return to crawling. There is a high risk for developing a relatively asymptomatic chronic uveitis, especially in ANA-positive individuals, requiring regular ophthalmology examinations to detect early changes. (See Table 107-3 for guidelines and “Uveitis” later for treatment.) Other complications that can be prevented in most patients with proper treatment include growth discrepancies in muscle tone and bulk, leg length, and the development of micrognathia and joint contractures.
Table 107-3 American Academy of Pediatrics Recommended Ophthalmologic Screening Frequency for Asymptomatic Uveitis in Juvenile Idiopathic Arthritis (JIA) Patients*67
Among children with oligoarthritis, 75% to 85% have a positive ANA (70% to 80% in persistent oligoarticular JIA and 80% to 95% in extended oligoarticular JIA),10 with low to moderate titers (1 : 40 to 1 : 320). The rate of ANA positivity is even higher in girls with an early onset.55 Patients are RF negative, although ACPAs have been detected in some patients with oligoarticular JIA, depending on the enzyme-linked immunoreceptor assay (ELISA) method used for screening.11 Some suggest that ANA serology should delineate a homogeneous group of arthritis patients, independent of the course of arthritis and number of joints involved.6
Differential Diagnosis
The differential diagnosis of oligoarticular JIA includes other JIA subtypes, especially ERA and psoriatic JIA; other rheumatic diseases of childhood; and nonrheumatic causes of joint pain and swelling such as septic arthritis, benign or malignant tumors, reactive arthritis, foreign body synovitis, pigmented villonodular synovitis, arterial-venous malformation, bleeding disorders (such as hemophilia), or severe trauma including nonaccidental injury. Mild trauma such as from a fall does not cause persistent joint swelling, and trauma is rarely a cause of joint swelling unless there is an internal derangement seen in older, not younger, children. Children with hypermobility can develop transient joint effusions after exercise,56 but this should not be long-lasting swelling. Lyme disease (in an endemic area) can cause recurrent monoarticular arthritis (typically involving the knee and often popliteal cysts), usually for less than 6 weeks. As described earlier, ALL may present with bone and joint pain and swelling, often monoarticular. If there is any concern of malignancy or infection, a complete blood count with manual differential and peripheral smear is crucial and bone marrow examination should be performed if indicated.
Treatment
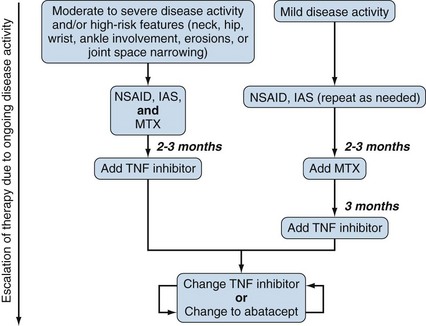
The treatment strategies for extended versus persistent oligoarticular JIA differ in both the approach to therapy and the intensity of escalating treatment on the basis of the number of joints at risk for significant damage. For extended oligoarticular JIA, treatment approaches are similar to RF-positive or RF-negative polyarticular JIA because these subtypes all have polyarticular involvement (see Figure 107-1). Treatment of persistent oligoarticular JIA is usually approached in a stepwise fashion (Figure 107-2). For these patients with a history of arthritis in four or fewer joints, the initial treatment is an NSAID, with or without an adjunctive IAS, followed by MTX if there is an inadequate response to one or more IASs. Initiation of MTX was recommended in the ACR guidelines for JIA as initial treatment for children with oligoarticular JIA with high disease activity and poor prognostic features, defined as involvement of hip, cervical spine, ankle or wrist, high inflammatory markers, or radiologic changes.9 Partial or complete remission on MTX can be induced in 60% to 70% with extended oligoarticular JIA.15,57,58 TNF inhibition should be considered in resistant cases, often in combination with MTX.
Special Considerations: Knee Monoarthritis
Because knee monoarthritis is the most common presentation of oligoarticular JIA, specific management is discussed here. The two treatments to be considered are NSAIDs and IASs, with evidence that IASs may be more effective even though most pediatric rheumatologists use NSAIDs before IASs.59 An initial trial of NSAIDs may be conducted with the hopes of avoiding IASs in some patients, but the risk of IASs must be weighed against the cost of continued active arthritis. Choosing among treatment strategies involves a tradeoff between several different outcomes including duration of active arthritis, potential for long-term complications, adverse effects of therapies, discomfort of daily medications or potentially painful procedures and anesthesia, as well as parental preferences. Synovectomy is not indicated in oligoarticular JIA.
Outcome
Long-term studies of adults treated before the use of biologic agents have shown that up to 50% of adults who had oligoarticular JIA may have ongoing active disease or functional problems in adulthood,60 and the rate of remission after 6 to 10 years from onset of disease ranges only from 23% to 47%.51,61 Ongoing disease activity and extension of joint involvement is related to a poor outcome and radiographic damage,62 and therefore emphasis on control of disease activity is critical. Morbidity from long-term inflammation can cause problems as well such as leg-length discrepancy with knee arthritis, muscle atrophy, bony overgrowth, and joint contractions (Figure 107-3), as well as other growth abnormalities such as micrognathia.
Patients with persistent oligoarthritis generally have the best outcome with 68% of patients achieving clinical remission off medication.43,51 Patients with an extended oligoarticular JIA have higher cumulative duration of active arthritis,43 more erosive disease, and a higher risk of chronic disability.51 Only 31% of children with extended oligoarticular JIA achieved remission after discontinuing their medication.43 Relapses occur, and within 2 years after clinical remission off medication, flares occurred in 47% of patients with persistent oligoarthritis and 67% of patients with extended oligoarthritis.43 Diligence regarding ophthalmologic screening for asymptomatic uveitis needs to continue as described in “Uveitis.”
Uveitis
Clinical Manifestations and Diagnostic Features
Chronic anterior uveitis, defined as inflammation involving the anterior uveal tract including the iris and ciliary body, is the most frequent extra-articular manifestation of JIA. Uveitis is most common in oligoarticular JIA and RF-negative polyarticular JIA, with a prevalence ranging from 17% to 26% and 4% to 25%, respectively. It is rare in RF-positive polyarticular JIA and sJIA.10 Risk factors for uveitis include ANA positivity, being younger than 6 years of age at JIA onset, female sex, and oligoarticular subtype.63 These risk factors of developing uveitis are larger influences in girls but not boys.64 The interval from diagnosis of JIA to the development of uveitis is longer the younger the age at onset of JIA, especially in ANA-positive patients.64 Overall, uveitis is observed in 30% of ANA-positive patients with JIA.65
Chronic anterior uveitis is typically nongranulomatous and asymptomatic at onset, and if unrecognized it can lead to serious visual deficits. Because of its insidious onset, regularly scheduled screening by an experienced ophthalmologist with slit lamp examination is required for early diagnosis and treatment. Newly diagnosed patients are ideally screened within 6 weeks of diagnosis66 because in 5% of cases, uveitis occurs before diagnosis of JIA.67 The highest risk period for uveitis development is the first 4 years after arthritis onset, although the risk is never completely eliminated.67 The frequency of ophthalmologic screening according to the American Academy of Pediatrics guidelines is determined by the degree of risk such as ANA status, age of JIA onset, and duration of disease shown in Table 107-3.67 In children with HLA-B27–related disease, anterior uveitis occurs in 10% to 15% but is usually highly symptomatic and therefore does not require routine screening.63 The severity of chronic anterior uveitis associated with JIA is unrelated to the severity of the underlying joint disease, and the clinical course of the uveitis and arthritis may not parallel one another. Disease is eventually bilateral in nearly two-thirds of patients, but both eyes are not always inflamed at the same time.68
Treatment
Differential Diagnosis
When evaluating the etiology of uveitis, it is important to consider infectious causes, such as tuberculosis, toxoplasmosis, cytomegalovirus, herpes simplex virus, syphilis, human immunodeficiency virus, Lyme disease, cat scratch disease, and fungus. Uveitis can be confined primarily to the eye or occur secondary to a systemic illness, such as in Behçet’s disease, sarcoidosis, autoinflammatory syndromes, multiple sclerosis, TINU (tubulointerstitial nephritis and uveitis syndrome), and Vogt-Koyanagi-Harada syndrome. Although the cause of uveitis often remains idiopathic, it is important to not miss a malignancy such as lymphoma or retinoblastoma.63
Uveitis is initially treated with topical corticosteroids and mydriatics, although in approximately 30% of patients the uveitis remains active even with topical or local treatment with subtenon corticosteroid injections, and immunosuppressive medications are indicated to attempt to aggressively control the inflammation and prevent poor visual outcomes. Although there are no prospective randomized controlled studies on the use of immunosuppressive medications in children with uveitis, several observational studies suggest the effectiveness of these treatments. MTX, either oral or parenteral, is usually the first choice of treatment and is also used as a steroid-sparing agent.69–71 Other effective immunosuppressives include mycophenolate mofetil72 and azathioprine, while cyclosporine has limited value.73 Treatment of uveitis requires close collaboration between the affected child’s rheumatologist and ophthalmologist.
In cases of refractory inflammation, biologic agents should be considered. TNF inhibitors are the most commonly used, specifically infliximab and adalimumab, which appear to be more effective than etanercept.74–81 The dosing range and frequency of infliximab varies widely when used for uveitis, and up to 10 mg/kg/month are often required for control and should be used with MTX to prevent development of anti-infliximab antibodies. Adalimumab is given 20 to 40 mg subcutaneously every 2 weeks, and when ineffective, it can be administered weekly.78 A case series reported remission with the administration of abatacept 10 mg/kg intravenously monthly in six of seven patients refractory to immunosuppressives and TNF inhibition.82 High-dose intravenous daclizumab, a humanized monoclonal antibody against IL-2 receptor, has been reported to be effective but is no longer available in the United States.83
In the past, ophthalmologists were concerned about primary placement of intraocular lens in JIA patients with history of uveitis with subsequent formation of cataracts.84 Now, with the more widespread practice of strict control of uveitis, good visual outcomes with cataract surgery and intraocular lens placement can be achieved using aggressive systemic immunomodulatory therapy perioperatively.85 The general expert opinion among uveitis specialists is to try to taper immunomodulatory therapy after 12 to 24 months of quiescence of uveitis; however, this has not been studied.86

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


