61 Traditional DMARDs
Methotrexate, Leflunomide, Sulfasalazine, Hydroxychloroquine, and Combination Therapies
Methotrexate
Key Points
MTX is polyglutamated in cells, and this is responsible for its long therapeutic effect.
Concomitant use of folic acid abrogates some of the side effects of MTX without decreasing efficacy.
The dose of MTX must be adjusted for reduced renal function.
Although rare, MTX pneumonitis is a serious and potentially fatal complication of therapy.
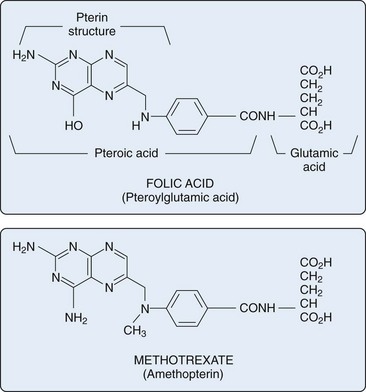
Chemical Structure
MTX is a structural analogue of folic acid and has substitutions in the pteridine group and para-aminobenzoic acid structure (Figure 61-1). The structure of folic acid (pteroylglutamic acid) consists of three elements: a multi-ring pteridine group, linked to a para-aminobenzoic acid, which is connected to a terminal glutamic acid residue.
Actions of Methotrexate
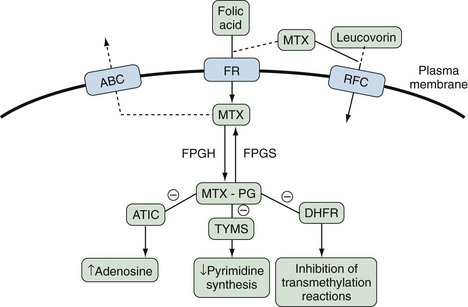
Because MTX is a folate analogue, it enters cells via a reduced folate carrier (RFC). Leucovorin competes with MTX for uptake using the same RFC; however, folic acid enters cells via another group of transmembrane receptors called folate receptors (FRs).1 FRs may be upregulated in cells with increased metabolic activity, including synovial macrophages, and serve as a second conduit for MTX influx.2,3 MTX efflux occurs via members of the adenosine triphosphate (ATP)-binding cassette (ABC) family of transporters, specifically ABCC1-4 and ABCG2.4 Genetic polymorphisms may affect MTX transporter proteins (influx and efflux) and can result in a variable MTX response and toxicity profile.4 Furthermore, multidrug resistance proteins have been identified that transport MTX, folic acid, and leucovorin out of cells, leading to MTX resistance.5
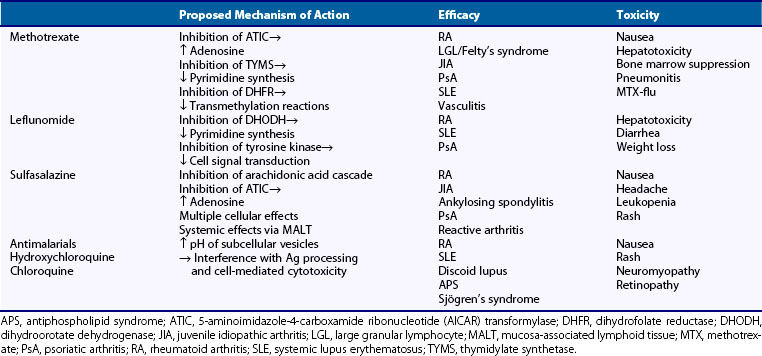
Once inside the cell, naturally occurring folates as well as MTX undergo polyglutamation by the enzyme folyl-polyglutamyl synthetase (FPGS). Polyglutamation of MTX (MTX-PG) is essential to prevent efflux of MTX, which easily occurs in the monoglutaminated state. MTX-PG has several key inhibitory effects on intracellular enzymes, which result in its postulated anti-inflammatory and antiproliferative (immunosuppressive) mechanisms: (1) Inhibition of aminoimidazole carboxamide ribonucleotide (AICAR) transformylase (ATIC) results in increased intracellular and extracellular adenosine, (2) inhibition of thymidylate synthetase (TYMS) results in decreased pyrimidine synthesis, and (3) inhibition of dihydrofolate reductase (DHFR) results in inhibition of transmethylation reactions essential for cellular functioning (Figure 61-2).
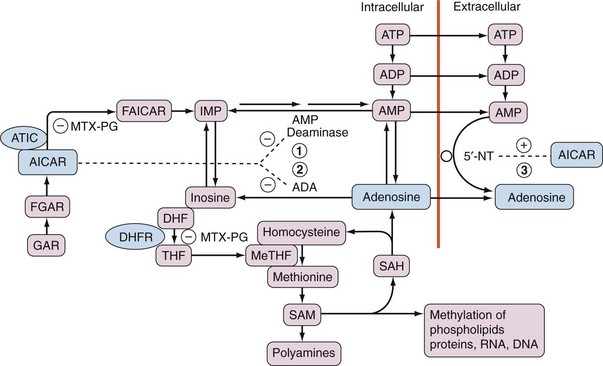
Inhibition of ATIC by MTX-PG leads to accumulation of AICAR and ultimately to increased levels of adenosine. Three possible mechanisms are postulated and likely work in combination: (1) AICAR inhibition of adenosine monophosphate (AMP) deaminase leads to excess production of adenosine from AMP; (2) AICAR inhibition of adenosine deaminase (ADA) leads to decreased breakdown of adenosine to inosine; and (3) AICAR stimulation of the ecto-5′-nucleotidase converts extracellular AMP to adenosine6–8 (Figure 61-3).
Adenosine, a purine nucleoside, has been termed a “retaliatory metabolite” because of its tissue protective functions after stressful injurious stimuli.9 Adenosine, a potent inhibitor of inflammation,9 induces vasodilation.10,11 Adenosine’s anti-inflammatory effects include regulation of endothelial cell inflammatory functions, including cell trafficking,10,11 counterregulation of neutrophils and dendritic cells,9,12 and cytokine modulation of monocytes and macrophages.9 Adenosine receptor ligation on monocytes and macrophages suppresses interleukin (IL)-12, a strong proinflammatory cytokine.13 Adenosine also suppresses the proinflammatory mediators tumor necrosis factor (TNF), IL-6, IL-8, macrophage inflammatory protein (MIP)-1α, leukotriene (LT)B4, and nitric oxide and enhances production of the anti-inflammatory mediators IL-10 and IL-1 receptor antagonist.14–19 Furthermore, adenosine receptor–mediated processes result in inhibition of the synthesis of collagenase, including tissue inhibitors of metalloproteinases.20 In sum, adenosine appears to promote a self-limiting, healthy immune response, hastening the transition from neutrophil-mediated inflammation to a more efficient and highly specific dendritic cell–mediated response. Ultimately adenosine leads to the resolution of inflammation by downregulation of macrophage activation and promotes a shift from a T helper (Th)1 cell to a T helper (Th)2 cell response.9
Evidence that the anti-inflammatory effects of MTX are mediated through adenosine has accumulated in in vitro and in animal studies.21 However, owing to adenosine’s short blood half-life of 2 seconds and MTX’s long latent period for active metabolites that modulate adenosine, it has been difficult to demonstrate changes in blood adenosine levels directly related to MTX.22 Recent evidence using forearm blood flow as a surrogate marker for adenosine release in RA patients treated with MTX demonstrated that MTX inhibits deamination of adenosine and potentiates adenosine-induced vasodilation.23 Demonstration of altered adenosine kinetics in patients treated with MTX coupled with adenosine’s known anti-inflammatory effects lends further credence to the hypothesis that MTX increases extracellular adenosine, which likely mediates some of the anti-inflammatory effects of MTX.
In addition to vasodilation, adenosine’s cardiovascular effects include negative inotropic and chronotropic cardiac effects, inhibition of vascular smooth muscle cell proliferation, presynaptic inhibition of sympathetic neurotransmitter release, and inhibition of thrombocyte aggregation.24 RA patients have a higher incidence of cardiovascular disease than the general population.25 MTX has been suggested to have a preferentially beneficial effect on cardiovascular mortality compared with other DMARDs in RA, and this effect is likely via adenosine modulation.26
The anti-inflammatory and antiproliferative effects of MTX may be mediated through its inhibition of transmethylation reactions. Both MTX and MTX-PG inhibit DHFR, resulting in diminution of tetrahydrofolate (THF). THF acts as a proximal methyl donor for several reactions by donating the methyl group for the conversion of homocysteine to methionine. Methionine is then converted to S-adenosylmethionine (SAM), which acts as a methyl donor for the following: methylation of RNA, DNA, amino acids, proteins, and phospholipids, and synthesis of the polyamines spermidine and spermine. Upon demethylation of SAM to S-adenosylhomocysteine (SAH), SAH is converted to adenosine and homocysteine. Methylation products that are dependent upon SAM, and thus indirectly upon DHFR, to generate THF are required for cellular survival and function, although specific cellular dependence upon each varies17 (see Figure 61-3).
The role of polyamines deserves further discussion. Spermine and spermidine have been shown to accumulate in urine,27 in peripheral blood mononuclear cells,28 and in synovial fluid and tissue29 in patients with RA. Metabolism of polyamines by mononuclear cells gives rise to toxic agents, including ammonia and hydrogen peroxide, which may impair lymphocyte function.30,31 Additionally, accumulation of polyamines in B cells is associated with enhanced production of rheumatoid factor (RF) in vitro, and incubation of these cells with methotrexate diminishes their ability to secrete both immunoglobulin and RF.17 These effects are seen with high in vitro concentrations of MTX and may not translate into the in vivo therapeutic effects of MTX in RA.
In addition, MTX inhibits methylation of 2′-deoxyuridylate (dUMP) into 2′-deoxythymidylate (dTMP) by TYMS, resulting in a further mechanism for disruption of DNA synthesis and proliferation of anti-inflammatory cells. This effect has been shown in vitro in human peripheral blood mononuclear cells incubated with low concentrations of MTX.32 Cell cycle disruption may lead to apoptosis of mononuclear cells via CD95 (APO-1/Fas) ligand-dependent33 and -independent mechanisms.34
Treatment with MTX has been shown to modulate monocytic and lymphocytic cytokines and their inhibitors. MTX has been shown to inhibit proinflammatory cytokine IL-1 secretion and to induce the IL-1 receptor antagonist, effectively inhibiting cellular responses to IL-1.35,36 Soluble TNF receptor (sTNFR p75) synthesis upregulation has also been shown as a result of MTX treatment from cultured monoblastic leukemia cells, which results in a diminished TNF inflammatory effect.37 MTX also inhibits production and secretion of the proinflammatory cytokine, IL-6, by cultured human monocytes.38,39 Reverse transcriptase polymerase chain reaction has been used to study the effects of MTX on gene expression for lymphocytic cytokines.40,41 MTX increases anti-inflammatory Th2 cytokine (IL-4 and IL-10) gene expression and decreases proinflammatory Th1 cytokine (IL-2 and interferon [IFN]-γ) gene expression in peripheral blood mononuclear cells (PBMCs) of patients with RA.41
Prostaglandins (PGs) and leukotrienes (LTs) are important mediators of joint destruction in RA. MTX has been shown to modulate the inflammatory enzymes cyclooxygenase (COX) and lipoxygenase (LOX), and their products PG and LT. Thromboxane B2 and prostaglandin E2 activities were reduced in the whole blood of RA patients treated with MTX when compared with healthy controls.42 MTX also reduces LTB4 synthesis by neutrophils, resulting in a decrease in total plasma LTB4 levels in patients with RA treated weekly with MTX.43 In addition to possible direct effects on COX and LOX, MTX has been shown to exert an inhibitory effect on neutrophil chemotaxis, which may result in a further reduction of these enzymes in sites of inflammation.44
Tissue destruction at sites of inflammation is thought to be related to increased synthesis and activity of proteolytic enzymes released by inflammatory cells, particularly in RA. MTX treatment has been shown to reduce gene expression of collagenase, metalloproteinase-1, and stromelysin, and to upregulate expression of tissue inhibitor of metalloproteinase-1 (TIMP-1).45 MTX may exert direct effects on messenger RNA (mRNA) for certain enzymes, such as collagenase. MTX also likely exerts indirect effects on gene expression via upstream cytokine modulation (IL-1 and IL-6), in the case of matrix metalloproteinase (MMP)-1 and TIMP-1.46
Pharmacology
Absorption and Bioavailability
At low doses, MTX can be administered either orally or parenterally (subcutaneous or intramuscular), and absorption is rapid, peaking at 1 to 2 or 0.1 to 1 hour, respectively. The absorption of low-dose oral and parenteral MTX (<15 mg/wk) is roughly equivalent, but once the oral dose exceeds 15 mg/wk, absorption diminishes by as much as 30%.47 Absorption is not reduced by concomitant food intake, except for milk, which may be inhibitory,48 but may be reduced in the setting of intestinal pathology, such as inflammatory bowel disease or malabsorptive conditions.
Orally administered MTX is absorbed via the GI tract and passes through the liver via the portal vein; parenterally administered MTX passes through the liver via the hepatic artery. Although not prospectively studied in RA patients receiving long-term MTX treatment, the parenteral route should have diminished potential for hepatotoxicity. This effect has been seen in a retrospective study wherein more elevations in transaminases were noted when oral MTX was administered to the same individuals versus when given parenterally.49 A recent study at equivalent MTX doses showed improved efficacy in RA clinical end points for parenteral over orally administered MTX.50
Forty-one RA patients who received 10 mg/m2 of oral MTX had a mean bioavailability of 70% with a range of 40% to 100%.51 The mean absorption time was 1.2 hours. Four hours after MTX administration, synovial fluid concentrations equal serum levels.51 A recent study of high-dose oral MTX (median dose, 30 mg/wk) has shown that mean bioavailability is improved by splitting the dose by 8 hours compared with one single dose (0.90 and 0.76, respectively).52 The pharmacokinetics of subcutaneous MTX are equivalent to those of intramuscular MTX; maximum serum concentration is attained within 2 hours of injection by either route.53 Also, bioavailability is equivalent between tablets and orally administered parenteral solution.54
Distribution and Half-Life
MTX is 50% to 60% bound to plasma proteins and has a half-life of approximately 6 hours.51 An increase in free MTX caused by displacement from albumin by more highly protein-bound drugs such as aspirin, nonsteroidal anti-inflammatory drugs (NSAIDs), and sulfonamides can occur. This is generally of limited clinical significance with low MTX doses because the increase in free MTX is usually only modest.
MTX accumulates in third space fluids, which can serve as a reservoir for redistribution into the circulation long after the last dose is administered.55 Caution should be used when administering MTX to patients with pleural effusions or ascites. Furthermore, unexpectedly high levels of MTX have been seen in patients with bladder cancer who have undergone ileal conduit surgery owing to enhanced intestinal absorption through the newly fashioned conduit.56
The biologically active form of MTX occurs after intracellular polyglutamation (MTX-PG). MTX undergoes up to five polyglutamations, and recent studies have looked at this aspect of MTX pharmacology. Once on a stable dose of MTX, the median time until 90% of the maximum steady-state concentration of MTX-PG was reached was found to be 27.5 weeks (range, 6.6 to 62.0 weeks).57
Elimination
Most MTX is excreted in the urine within the first 12 hours after administration, except for MTX-PG. MTX undergoes some hepatic metabolism by the enzyme aldehyde oxidase to the 7-hydroxymethotrexate metabolite; this metabolite has unknown significance in RA. MTX and metabolites are excreted by the kidney by glomerular filtration and proximal tubular secretion but also undergo distal tubular reabsorption. The estimated median half-life of elimination of MTX-PG is 3.1 weeks (range, 0.94 to 4.1 weeks), and MTX-PG is undetectable at 15 weeks.57 MTX-PG3 is the most common subtype seen (30% of total MTX-PG) and has a median half-life elimination of 4.1 weeks.
Indications
Rheumatoid Arthritis
The efficacy of MTX in RA has been clearly established. Four well-designed, blinded, placebo-controlled trials58–61 published in 1984 and 1985 had a tremendous impact on the treatment of RA. These trials varied in design and duration: Two of these trials used oral MTX and two used intramuscular (IM) MTX, two trials had a crossover and two were parallel, and the duration of treatment varied from 6 to 28 weeks. Although the design and duration of therapy in these trials varied, the conclusions did not, as all showed MTX to be superior to placebo in the short-term treatment of RA. A meta-analysis of these trials by Tugwell and coworkers62 showed that MTX-treated patients had a 37% greater improvement in swollen joint and tender joint scores, a 39% greater improvement in joint pain, and a 46% greater improvement in morning stiffness. MTX was generally well tolerated in these trials; withdrawal rates ranged from 0 to 32% and were mostly related to minor toxicities (i.e., stomatitis, nausea). Taken together, the results of these trials firmly established MTX as an effective therapy for the treatment of RA, at least in the short term.
Numerous trials have compared MTX with other DMARDs. A meta-analysis done by Felson and co-workers showed that MTX was superior to placebo, auranofin, and probably hydroxychloroquine (HCQ), and was comparable with penicillamine, sulfasalazine, and IM gold.63 No trial has ever suggested that any other synthetic DMARD is superior to MTX.
Accumulating evidence suggests that the short-term benefit of most DMARDs is not sustained, and few patients continue to take these drugs after 3 years.64,65 MTX appears to have the best durability. Pincus and colleagues have shown that 60% of patients continued MTX at 5 years, compared with less than 25% for penicillamine, gold, HCQ, and azathioprine.64 Of all the DMARDs, MTX appears to have the best efficacy-to-toxicity ratio.63 However, despite all the favorable efficacy reports, MTX alone rarely induces remissions of RA, and it has become the cornerstone of combinations of DMARD therapies as discussed later.66,67
Rheumatoid Arthritis–Related Conditions
MTX has been used successfully in treating Felty’s syndrome68 and the large granular lymphocyte syndrome when it is found in patients with RA.69 Improvement in neutrophil count occurs within 4 to 8 weeks of MTX initiation in both cases. MTX has been used successfully in adult-onset Still’s disease70 and for the cutaneous vasculitis of RA.71
Juvenile Idiopathic Arthritis
MTX is efficacious in juvenile idiopathic arthritis. A definitive, randomized, placebo-controlled trial demonstrated that MTX at a dose of 10.0 mg/m2 was superior to 5.0 mg/m2 or placebo.72 Sixty-three percent of children receiving the higher dose (10.0 mg/m2) of MTX improved, compared with 32% in the lower-dose group (5.0 mg/m2) and 36% in the placebo group.
Psoriatic Arthritis (PsA)
Numerous prospective and retrospective trials have showed a benefit for MTX in PsA.73 The largest double-blind randomized trial compared weekly oral MTX with placebo, and showed statistically significant results only for physician global assessment of arthritis activity and the amount of affected skin surface area; however, this study was small and may have been underpowered to detect differences in joint count, pain, and swelling.74 Despite the paucity of randomized controlled trial data, MTX remains a commonly used systemic agent in the treatment of PsA.
Systemic Lupus Erythematosus (SLE)
MTX has been shown to be efficacious in controlling cutaneous and/or articular manifestations of SLE, particularly in disease resistant to antimalarials or requiring high doses of systemic steroids.75 Concomitant folic acid should be administered to abrogate the elevated levels of homocysteine that may be a side effect of MTX therapy, and is considered a risk factor for cardiovascular disease in SLE. The role of MTX in treating more severe SLE involvement, including renal, hematologic, or central nervous system disease, has not yet been established.76 Extreme caution should be employed in patients with renal disease.
Vasculitis
MTX in conjunction with corticosteroids has shown efficacy in treating early and non–life-threatening granulomatosis with polyangiitis (formerly Wegener’s granulomatosis), including upper airway disease and mild renal disease.77–80 In addition to induction of remission, MTX has been shown to maintain remission in granulomatosis with polyangiitis, although vigilance for relapse is warranted.81 Despite a lack of well-designed studies, MTX has been shown to be efficacious in corticosteroid-resistant Takayasu’s arteritis82 and in relapsing polychondritis.83
The use of MTX for polymyalgia rheumatica (PMR) and giant cell arteritis (GCA) has been controversial. Multiple open-label studies have shown differing results. Recently, randomized, placebo-controlled trials (RCTs) of MTX in addition to corticosteroids in PMR and GCA have shown conflicting results.84–87 Thus, the routine use of MTX in either PMR or GCA has not been adopted, but some advocate for its use in an effort to more rapidly taper corticosteroid in those patients with intolerable side effects.
Inflammatory Myopathies
A review of the published reports of MTX and the inflammatory myopathies polymyositis (PMS) and dermatomyositis (DMS) shows overall positive results.88 However, despite the frequent use of MTX in the inflammatory myopathies, a recent Cochrane database review reveals a paucity of well-designed trials.89
Other Rheumatic Diseases
MTX has been used in systemic sclerosis. One RCT looking at MTX use in early systemic sclerosis90 showed a trend in benefit for skin scores and pulmonary diffusion capacity and a significant benefit for physician global assessment; a second RCT in established systemic sclerosis91 showed significant benefit for skin scores and total creatinine clearance. In addition, prospective trials have shown that MTX is efficacious in the treatment of corticosteroid-resistant multisystem sarcoidosis,92,93 and a recent RCT showed that if initiated early in sarcoidosis, MTX is an effective steroid-sparing agent.94 MTX has also been shown to be effective as primary treatment and as a corticosteroid-sparing agent in inflammatory ocular disease.95,96 Finally, MTX in combination with corticosteroids is effective in the treatment of multicentric reticulohistiocytosis.97
Dose and Drug Administration
MTX is available as 2.5, 5, 7.5, 10, and 15 mg tablets, and as a solution of 25 mg/mL for subcutaneous or intramuscular injection. The starting dose is usually 5 to 10 mg given as a single weekly dose. More frequent administration is associated with a significantly increased risk of liver toxicity.98 If the oral dose of MTX exceeds 15 mg, consideration should be given to splitting the dose, with each half given 6 to 12 hours apart, for improved bioavailability. The dosage of MTX can be escalated, usually every 4 to 8 weeks to 25 mg/wk to achieve the desired clinical response. MTX may be administered orally via tablet or parenteral solution; the latter is less costly. Because its bioavailability is variable and appears to decrease at higher doses, parenteral MTX is generally recommended if patients have active disease despite oral doses of approximately 20 mg/wk. Recent data have shown that parenteral MTX is clinically superior to orally administered MTX, and that this may be a preferred route of administration, especially if oral MTX is not optimally effective.50
Concomitant administration of folic acid (1 to 3 mg/day) decreases the frequency of toxicities, including mucositis, nausea, hematologic abnormalities, and liver enzyme elevations, without seeming to interfere with clinical efficacy.99,100 Folic acid administration also decreases hyperhomocysteinemia in patients on MTX, and this may be important to help decrease the already high cardiovascular risk of patients with RA. Low-dose folinic acid has also been used and can markedly reduce MTX toxicity in rheumatic disease therapy without interfering with efficacy if given in doses of 2.5 to 5 mg/wk and if not administered until 24 hours after the MTX dose. Because folic acid is widely available and less expensive, it is preferred by most.
Measurement of MTX-PG levels is commercially available (MTXGlun). It would be valuable to have a marker to predict response and adverse events associated with MTX therapy, but mixed results have been obtained in the search for a trend in the dose-response relationship for MTXGlun levels and RA disease activity. A recent study showed no relationship between MTXGlun concentration and reduced disease activity in RA.101 Furthermore, no relationship was identified between MTXGlun levels and adverse events. Disease activity was influenced by red blood cell (RBC) folate level, and further study is warranted to determine whether this may serve as a marker for MTX efficacy.
Geriatric Patients
Patients older than 65 years represent a special subset of patients receiving pharmacotherapy. Pharmacokinetic profiles, including drug distribution, are changed in the elderly as the result of decreases in end-organ blood flow and lean body mass, decreased hepatic drug metabolism, and decreased renal drug excretion. Furthermore, these patients are more likely to have multiple comorbidities, polypharmacy, noncompliance, increased risk for dosage errors, and limited access to medication for financial reasons.102
In practice, recommended doses should be reduced when therapy is initiated and should be adjusted for renal function based on creatinine clearance (CrCl).103 The serum creatinine may be a misleading measure of renal function in older patients owing to an overall reduction in lean muscle mass. Dosing recommendations are as follows: Initial doses should be around 5 to 7.5 mg/wk and should not exceed 20 mg/wk. Dosage adjustments for CrCl are as follows: For a CrCl of 61 to 80 mL/min, reduce the dose by 25%; for a CrCl of 51 to 60 mL/min, reduce the dose by 30%; for a CrCl of 10 to 50 mL/min, reduce the dose by 50% to 80%; and for a CrCl less than 10 mL/min, avoid use104 (Table 61-1).
Toxicity
Gastrointestinal and Hepatic Side Effects
Gastrointestinal symptoms, including dyspepsia, nausea, and anorexia, are common, occurring in up to 20% to 70% of patients within the first year of therapy.105 These symptoms may be attenuated by adding folic acid or by changing to a parenteral dosing regimen.
The risk of significant liver toxicity appears to be low when MTX is given once weekly to patients who abstain from alcohol consumption and are monitored carefully, and is on the order of 1 case per 1000 after 5 years of use.106 However, a recent review of a large North American database of patients with RA and PsA found that elevations in aspartate aminotransferase (AST)/alanine aminotransferase (ALT) greater than 1 times the upper limit of normal (1 × ULN) occurred in 22% and 31% of patients taking MTX or MTX and leflunomide. Elevations greater than 2 × ULN occurred in 1% to 2% on monotherapy and in greater than 5% on combination therapy. Elevated liver function tests (LFTs) were more likely in patients with PsA.107 Alcohol consumption, α1-antitrypsin deficiency, morbid obesity, diabetes, concomitant hepatotoxic drugs, and chronic hepatitis B or C have all been implicated as possible risk factors for MTX toxicity.108
Hematologic Side Effects
Bone marrow toxicity, in most cases, is dose dependent and responds to folic acid administration. Pancytopenia, leukopenia, anemia, and thrombocytopenia can occur, but are rare. In a review by Gutierrez-Urena and associates, clinically significant pancytopenia was found to develop in up to 1% to 2% of RA patients on MTX therapy.109 Severe, life-threatening bone marrow toxicity can be treated with folinic acid (leucovorin) and, if necessary, granulocyte-stimulating factor (GSF). Because the elimination of MTX is dependent on the kidney, decreases in renal function may precipitate bone marrow toxicity in patients who have been previously stable. Additional risk factors include hypoalbuminemia, dosing errors, and concomitant use of probenecid or trimethoprim/sulfamethoxazole (TMP/SMX).
Pulmonary Side Effects
Five clinical pulmonary syndromes have been associated with MTX treatment: acute interstitial pneumonitis (hypersensitivity pneumonitis), interstitial fibrosis, noncardiogenic pulmonary edema (seen in high-dose treatment for malignancy with rare reports in RA), pleuritis and pleural effusions, and pulmonary nodules.110 Time lapse from initiation of therapy to cumulative dose before the onset of pulmonary toxicity is extremely variable, at 1 to 480 weeks and 7.5 to 3600 mg of MTX, respectively.110 MTX-induced pulmonary disease is rare and is difficult to quantify, but estimates suggest an incidence of 3.9 cases per 100 patient-years of MTX exposure and a prevalence of 2.1% to 5.5%.111,112
Patients generally present with shortness of breath, tachypnea, dry cough, and fever. Chest radiographs most typically show a bilateral interstitial infiltrate (although this varies). Infectious causes, including opportunistic organisms, must always be ruled out. If routine evaluations for infection, including sputum studies, and for other medical conditions to explain the pulmonary symptoms are negative, bronchoscopy with bronchoalveolar lavage and transbronchial biopsy is recommended. If MTX pulmonary toxicity is suspected, MTX should be discontinued and supportive treatment initiated with the use of corticosteroids in more severe cases. Some patients with pulmonary toxicity have been successfully restarted on MTX,113 but clinicians have reported mortality in up to 50% of retreated patients.55
Factors that appear to predispose to MTX lung toxicity include age, blue collar occupation, smoking (in women), diabetes, pleuropulmonary rheumatoid disease, and skin rashes from MTX.114
Malignancies
The induction of malignancies by MTX is a concern, and several studies have examined this question with conflicting conclusions. Recently, reports of lymphoma in MTX-treated RA patients have appeared. Because the incidence of lymphoma is increased in RA patients anyway,115 these reports are difficult to interpret. The case for a causative role of MTX has been strengthened, however, because a number of these cases have been B cell lymphomas of the type commonly seen in association with immunosuppression (associated with Epstein-Barr virus) and that may regress after discontinuation of MTX.116,117 Subsequently, lack of a causal relationship between MTX treatment and the development of lymphoma has been seen in two large series of RA patients—one prospective study115 and one retrospective study.118 The potential benefits of MTX for most RA patients thus far outweigh these statistically small risks.119
Miscellaneous
Nodulosis
The development of, or increase in, the number or size of rheumatoid nodules has been reported to occur in RA patients treated with MTX with a prevalence of up to 8%.120 This may occur in rheumatoid factor–negative patients and in those in whom the synovitis is under excellent control. The mechanism of this nodule formation has been suggested to be due to an increase in adenosine, which appears to promote nodule formation.121 Conversely, nodules have been reported to decrease during MTX therapy.
Fertility, Pregnancy, and Lactation
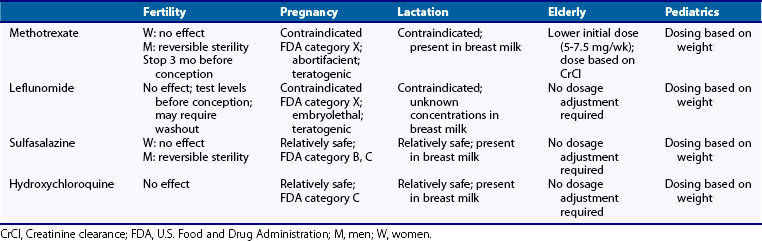
MTX does not seem to adversely affect female fertility but can cause reversible sterility in men.123 Women and men should discontinue MTX for at least 3 months before attempting to conceive, because of its large distribution and long half-life in the liver. Folic acid supplementation is essential before conception. MTX is included in U.S. Food and Drug Administration (FDA) Pregnancy Category X and is contraindicated in pregnancy. Women of childbearing age who are considered for MTX therapy should receive extensive counseling regarding teratogenic risk and should be placed and maintained on adequate contraception, before therapy is begun. Toxicities include fetal abnormalities such as “aminopterin syndrome” (multiple craniofacial, limb, and central nervous system abnormalities) and embryonic or fetal loss, and MTX at high doses (1 mg/kg) is an effective abortifacient. MTX is also contraindicated during lactation because small amounts are excreted in breast milk (see Table 61-1).
Toxicity Monitoring
The American College of Rheumatology (ACR) has recently revised recommendations for the use of DMARDs, and these serve as an excellent resource.124 Toxicities that require monitoring include myelosuppression, hepatotoxicity, and pulmonary toxicity. Baseline evaluation should include a complete blood count (CBC) with platelets, hepatitis B and C serology in high-risk patients, liver transaminases, and creatinine. Although these guidelines make no recommendations on the need for a baseline chest x-ray (CXR), this is a reasonable approach. Liver biopsies are not routinely recommended before MTX is initiated. The rare patients whom one wants to treat with MTX despite abnormalities in screening laboratory or other significant risk factors may require liver biopsy before MTX is initiated. In addition, biopsies are recommended only in those patients who continue to have enzyme abnormalities, and for whom continuation of MTX therapy is contemplated.
Monitoring for toxicity should be done every 2 to 12 weeks and is based on the duration of therapy, with more frequent monitoring provided earlier in the course of treatment. Systems review and physical examination should include monitoring for symptoms or signs of myelosuppression (fever, infection, bruising, and bleeding), pulmonary toxicity (shortness of breath, cough, rales), gastrointestinal intolerance (nausea, vomiting, diarrhea), and lymphadenopathy. Laboratory parameters that should be followed include a CBC with platelets, liver transaminases, and creatinine (Table 61-2).
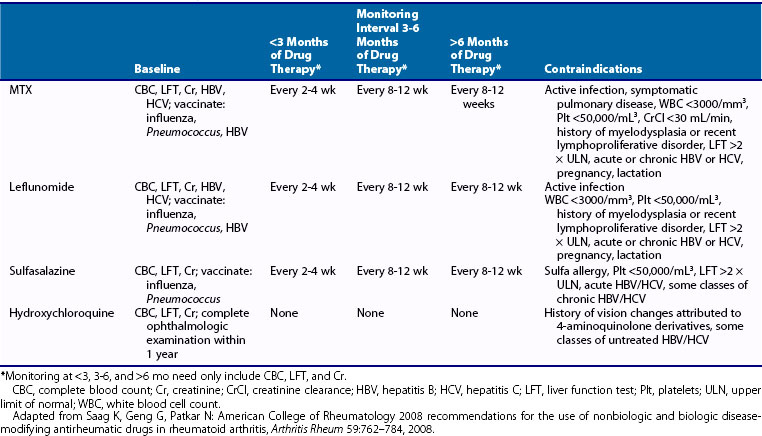
It is important to consider vaccination status in any patient who is going to use MTX. RA patients have an increased incidence of death from pneumonia,125 and MTX may reduce the immune response to pneumococcal antigen.126 Thus, any patient in whom MTX is going to be used should first receive the pneumococcal vaccination, with booster as appropriate. Vaccinations for hepatitis B virus for at-risk patients and yearly influenza vaccines are recommended as well. Caution should be exercised when administering live virus vaccinations to patients on MTX.
Drug Interactions and Contraindications
Drug Interactions
Drugs that are known hepatotoxins, such as sulfasalazine, leflunomide, and azathioprine, may potentiate liver toxicity when used in combination. Organic acids such as sulfonamides, salicylates, NSAIDs, penicillin G, piperacillin, and probenecid competitively inhibit tubular secretion, and this delays MTX clearance.127 MTX also undergoes distal tubular reabsorption, which may be enhanced by the addition of HCQ128 and blocked by the addition of folic acid.127 Drugs that affect renal function should be used with caution because of the renal clearance of MTX and, therefore, the increased risk of MTX toxicity that could occur because of decreased clearance.
Several of the aforementioned drugs deserve special mention. Trimethoprim-sulfamethoxazole should be avoided or used with extreme caution because of possible hematologic toxicity with MTX. Mechanisms for this toxicity include an additive antifolate effect from trimethoprim, decreased MTX clearance due to inhibition of tubular secretion by sulfamethoxazole, and altered MTX plasma protein binding. NSAIDs are commonly used in RA patients as adjunctive therapy. NSAIDs may increase MTX levels by displacing MTX from plasma proteins and limiting tubular secretion. Despite lack of a significant pharmacokinetic or clinical interaction between low-dose MTX and a variety of NSAIDs,129 vigilance for MTX toxicity should increase whenever NSAID dosages are changed in patients on stable weekly doses of MTX. Low doses of aspirin used for cardiovascular prophylaxis are not likely to be of concern. Furthermore, probenecid should be avoided because it inhibits tubular secretion of MTX.
Contraindications
MTX should not be used in severe renal, pulmonary, or hepatic impairment, pre-existing bone marrow suppression, alcoholic liver disease, and pregnancy or breastfeeding. Ongoing or active infection is also a contraindication. In most cases, patients who desire to continue drinking alcohol should not be treated with MTX. Mild to moderate renal insufficiency is a relative contraindication, and use of MTX in these patients may require more vigilant toxicity monitoring (Table 61-3).
Leflunomide
Key Points
Leflunomide reversibly inhibits dihydroorotate dehydrogenase (DHODH).
Loading doses often are not used in clinical practice because of gastrointestinal toxicity.
Because of enterohepatic recirculation, leflunomide has a very long half-life.
Vigilance must be used for hepatotoxicity.
Chemical Structure
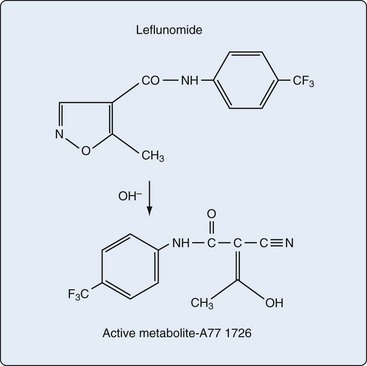
Leflunomide is a low-molecular-weight isoxazole compound and is chemically unrelated to any previous immunosuppressant. Leflunomide is a pro-drug and is rapidly and completely converted to its active metabolite, the malononitriloamide A77 1726. A77 1726 is also known as teriflunomide (Figure 61-4).
Actions of Leflunomide
As with MTX, the precise mechanism of action responsible for the effects of leflunomide in rheumatic disease is not completely understood.130 Leflunomide is immunomodulatory, with the net effect being a reduction in activated T lymphocytes. Its two in vitro mechanisms of action vary depending on concentration: (1) At the concentration of the active metabolite (A77 1726) achieved in patients, its major effect appears to be reversible inhibition of the enzyme dihydroorotate dehydrogenase (DHODH), which results in inhibition of pyrimidine synthesis; (2) at higher concentrations, A77 1726 also inhibits tyrosine kinases, interfering with cell signal transduction.131
Activation of T cells results in progression from the resting phase (G0) to the G1 phase, where ribonucleotides are synthesized, and then to the S phase, where cellular DNA is replicated in preparation for mitosis. T cell activation requires significant increases in de novo pyrimidine and purine biosynthesis. Sensors such as proto-oncogenes (p53) and checkpoints (cyclins C and D) in this pathway monitor the level of nucleotide pools and prevent damaged cells from replicating.131
Uridine monophosphate (rUMP) is a precursor for the formation of pyrimidine nucleotides and thus is essential for both RNA and DNA synthesis. The steps in de novo rUMP synthesis are seen in Figure 61-5. A critical step in this pathway is the generation of dihydroorotate in the cytoplasm with subsequent diffusion into the mitochondria, where the enzyme dihydroorotate dehydrogenase (DHODH) is located. DHODH converts dihydroorotate to orotate, and the latter diffuses back into the cytoplasm and is subsequently converted to rUMP and ultimately to RNA and DNA.
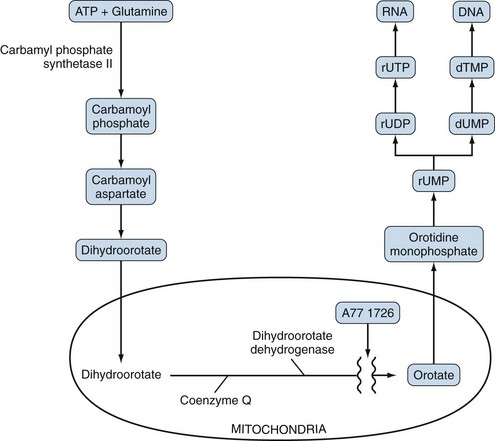
The first postulated mechanism of action of leflunomide consists of inhibition of DHODH by A77 1726, which lowers orotate levels and leads to a decrease in rUMP and subsequent nucleotide synthesis, resulting in T cell cycle arrest. This mechanism of action has been substantiated by experimental evidence. In vitro mitogen-stimulated activation of T cells is blocked by levels of A77 1726 that inhibit DHODH, and this inhibition can be reversed by the addition of uridine, suggesting that A77 1726 works by disruption of pyrimidine biosynthesis.132,133 Further, the only enzyme inhibited by A77 1726 in this pathway, at concentration obtained in vivo, is DHODH.134
Evidence also exists to support that inhibition of DHODH produces an arrest of lymphocytes in the G1 phase of the cell cycle.135 If the level of ribonucleotides, including rUMP, falls below a critical point, cytoplasmic p53 activation occurs, and p53 will translocate to the nucleus and initiate cellular arrest by ultimately preventing transcription of cyclins D and E. In cultures of human T cells, A77 1726 depletes rUMP pools and results in an accumulation of nuclear p53 with resultant cell cycle arrest.136 In comparison, treatment of cell lines lacking p53 with A77 1726 does not cause a G1 phase arrest.137
Resting lymphocytes maintain ribonucleotide requirements largely through salvage pathways and are essentially unaffected by leflunomide.138 Active, or autoimmune, lymphocytes rely on the de novo pathway and are affected by leflunomide. Over the course of treatment with this slow-acting agent, autoimmune lymphocytes should be removed progressively.131
At higher concentrations, A77 1726 inhibits phosphorylation of tyrosine kinases that are critical for cell growth and differentiation of activated cells.139,140 This inhibition has been proposed to partially or completely explain the antiproliferative effects of leflunomide; however, it is unclear whether concentrations sufficient to achieve this effect are obtained in vivo.
Several other additional anti-inflammatory properties of leflunomide have been noted. Leflunomide has the ability to block the activation of nuclear factor κB (NFκB),141 which regulates the expression of genes important in inflammatory processes, including those seen in inflammatory arthritis.142 Ex vivo and in vitro studies in humans have shown that both leflunomide and MTX inhibit neutrophil chemotaxis, which may decrease the recruitment of inflammatory cells into the joints.44 Leflunomide has also been shown to decrease the ratio of MMP-1 to TIMP-1.44 Finally, leflunomide alters the synthesis of cytokines by augmenting the immunosuppressive cytokine-transforming growth factor-β1 and suppressing the immunostimulatory cytokine IL-2.143
Pharmacology
Absorption and Bioavailability
The gastrointestinal tract and the liver rapidly and completely convert ingested leflunomide into A77 1726. Food does not interfere with absorption. Circulating A77 1726 is highly bound (more than 99%) to plasma proteins, predominantly albumin. Its plasma concentration is linearly correlated with a single oral dose over a range of 5 to 25 mg; steady state is reached in 7 weeks after daily dosing.144
Distribution and Half-Life
A77 1726 has a half-life of approximately 2 weeks (mean, 15.5 days),144 with a low apparent volume of distribution. A77 1726 undergoes enterohepatic recirculation. In healthy subjects, 90% of leflunomide is excreted by 28 days,144 but some may be present for much longer periods.
Elimination
In healthy subjects, the proportions excreted by the kidney and the gut are nearly equal. Because detectable A77 1726 may be present in the body months or years later, the ability to rapidly and effectively eliminate A77 1726 with cholestyramine is important. Oral administration of cholestryamine 8 g three times daily can lower the apparent half-life of A77 1726 to 1 to 2 days.145 Furthermore, activated charcoal 50 g every 6 hours can reduce plasma levels by 50% within 24 hours.145
Indications
Rheumatoid Arthritis
Leflunomide was first shown to be safe and effective in treating RA in a placebo-controlled, dose-ranging, 6-month trial.144 Two pivotal trials, one in Europe and one in the United States, have compared leflunomide with sulfasalazine (SSZ) and MTX. The European trial had three arms: leflunomide (20 mg/day after a loading dose), sulfasalazine (escalated to 2 g/day), and placebo.146 In this trial, both leflunomide and sulfasalazine were superior to placebo in terms of swollen and tender joint counts, as well as physicians’ and patients’ overall assessments. It is important to note that both the leflunomide and sulfasalazine groups reported significant effects on slowing radiographic progression of disease compared with placebo. The U.S. trial compared patients treated with leflunomide (20 mg/day after a loading dose), MTX (7.5 to 15 mg/wk), or placebo.147 Again, both active drugs were found to be superior to placebo but not different from each other. Both leflunomide and MTX also slowed radiographic progression of disease compared with the placebo group. Another trial compared leflunomide (20 mg/day with loading) with MTX (10 to 15 mg/wk) in a 1-year trial with a 1-year extension.148 In this trial, MTX was shown to be statistically superior to leflunomide for the clinical outcomes measured, as well as the rate of radiographic progression after 2 years.
Other Rheumatic Diseases
Leflunomide has been reported to be efficacious in systemic lupus erythematosus. In a randomized, controlled trial, leflunomide was more effective than placebo in improving markers of lupus disease activity, and was safe and well tolerated.149 A subsequent small, prospective, open-label trial of patients with lupus nephritis unresponsive to conventional therapy showed that leflunomide was efficacious and well tolerated.150
Leflunomide has been shown to be effective in treating psoriatic arthritis and psoriasis when compared with placebo.151 In an open-label trial of ankylosing spondylitis, leflunomide was shown to be effective in treating peripheral arthritis, but axial symptoms did not improve.152
Both an open-label trial153 and a randomized, controlled clinical trial154 have shown the efficacy of leflunomide in maintaining remission in granulomatosis with polyangiitis after successful induction with cyclophosphamide. In the latter trial, leflunomide was superior to methotrexate in preventing relapse.
Leflunomide has also been shown to be safe and effective in patients with juvenile idiopathic arthritis (JIA) who did not respond to or could not tolerate MTX.155
Dose and Drug Administration
Leflunomide is available in oral tablets at doses of 10, 20, and 100 mg. Oral leflunomide is rapidly metabolized to A77 1726, which has a very long half-life; therefore, the standard recommendation is to start therapy with a loading dose of 100 mg daily for 3 days, then switch to the standard maintenance dose of 20 mg daily. Despite this recommendation, many clinicians no longer prescribe a loading dose because it is believed to increase the drug’s gastrointestinal toxicity.156 Also, it is common practice to decrease the dose to 10 mg daily if toxicity occurs, or if complete control of the disease can be maintained. Because of its long half-life, some clinicians give leflunomide less often (three to five times per week).
Pediatric Patients (See Table 61-1)
Although not approved in the United States to treat patients with JIA, leflunomide is used off-label for this condition at a dose of 10 to 20 mg/day. This dosing is often based on a patient’s body weight. A recently published example of dosing by body weight is as follows: A patient weighing less than 20 kg receives 10 mg every other day, a patient weighing more than 20 kg but less than 40 kg receives 10 mg/day, and a patient weighing more than 40 kg receives 20 mg/day.157
Toxicity
For major controlled trials that have used leflunomide at a dose of 20 mg/day, the incidence of adverse events that resulted in trial withdrawal are shown in Table 61-4. Leflunomide-associated withdrawals (19%) were more frequent than those associated with MTX (14%), were similar in frequency to those associated with sulfasalazine (19%), but were more frequent than those associated with placebo treatment (8%).
Table 61-4 Leflunomide Trial Withdrawals for Adverse Events
| No. of Patients | Withdrawals | |
|---|---|---|
| Leflunomide | 816 | 154 (19%) |
| Methotrexate | 680 | 94 (14%) |
| Sulfasalazine | 133 | 25 (19%) |
| Placebo | 210 | 16 (8%) |
Gastrointestinal and Hepatic Side Effects
Liver toxicity can occur in association with leflunomide administration. Data from a large U.S. cohort of RA and PsA patients show that elevations in ALT/AST levels greater than 1 times the upper limit of normal (1 × ULN) occurred in 17% and elevations greater than 2 × ULN occurred in 1% to 2% of patients taking leflunomide. Leflunomide given in combination with MTX resulted in ALT/AST elevations greater than 1 × ULN in 31% and greater than 2 × ULN in 5% of patients. Furthermore, this change in transaminases was more commonly seen in PsA patients.107 The European Agency for Evaluation of Medicinal Products (EMEA) reported 296 patients with hepatic abnormalities and 15 patients with liver failure and death while taking leflunomide.158,159 The U.S. Food and Drug Administration (FDA) reviewed adverse event reports between August 2002, and May 2009, and found 49 cases of severe liver injury, 14 of which resulted in death.160 Most patients with hepatotoxicity have risk factors, including concomitant administration of another hepatotoxic agent or underlying liver disease.
Cardiovascular Side Effects
Hypertension has consistently been reported to occur more frequently in leflunomide-treated compared with placebo-treated patients.146,147 Additionally, elevation of cholesterol levels has been reported in association with leflunomide use.161 Both of these effects should be monitored because of the excess cardiovascular mortality reported in patients with RA.
Fertility, Pregnancy, and Lactation
It is critically important to note that the active metabolite of leflunomide (A77 1726), largely because of its enterohepatic circulation, may remain in the body for years. Therefore, if a woman who has previously received leflunomide wishes to become pregnant, A77 1726 levels should be measured. Active elimination of leflunomide from the body should be considered for levels above 0.02 mg/L. This can be achieved by the oral administration of cholestyramine for 11 days (8 g three times daily).163 Before pregnancy is attempted, verification of levels below 0.02 mg/L should be confirmed on two separate occasions, at least 14 days apart, and women should then wait an additional three full menstrual cycles.164 Patients may require more than one course of cholestyramine to achieve this level. Although no data exist, men wishing to father children should undergo the same washout procedure as women and should wait an additional 3 months after the second drug plasma level is verified below 0.02 mg/L (see Table 61-1).
Toxicity Monitoring
Similar to MTX, the ACR has published guidelines on the initiation and monitoring of leflunomide.124 Patients taking leflunomide should have a baseline CBC and liver enzyme monitoring, including AST, ALT, and albumin. Serum creatinine is important because leflunomide is partially eliminated by the kidney. The frequency of monitoring depends on the duration of therapy (see Table 61-2). More frequent monitoring may be warranted if concomitant immunosuppressive agents, such as MTX, are given. If patients experience significant toxicity, a washout procedure is indicated to more rapidly eliminate the drug. Caution should be exercised when live virus vaccinations are administered to patients on leflunomide.
Drug Interactions and Contraindications
Contraindications
Leflunomide should not be used in patients with impaired liver function, severe renal impairment, bone marrow dysplasia, severe immunodeficiency, severe hypoproteinemia, or known hypersensitivity to the drug. The liver is involved in enterohepatic recirculation and biliary excretion, thus leflunomide use in liver disease is contraindicated. In renal insufficiency, the levels of circulating A77 1726 do not appear to be increased, but the component of free A77 1726 is increased. Leflunomide is contraindicated in the setting of serious infection and should be discontinued in patients with new or worsening pulmonary symptoms or rash. Leflunomide is absolutely contraindicated in pregnancy and breastfeeding (see Table 61-1).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


