73 Sjögren’s Syndrome
Historical Perspective
In 1888 Johann von Mikulicz-Radecki described a case of bilateral painless swelling of the lacrimal, parotid, and submandibular glands,1 an entity that later bore his name. Reports soon followed showing Mikulicz disease was not a distinct pathologic entity but rather a clinical potpourri of conditions including leukemia, lymphoma, and tuberculosis. Shortly thereafter, salivary gland disease became linked with dryness of the eyes and oral mucosa when Henri Gougerot, a well-known and successful French dermatologist, wrote in 1925 about three cases of salivary gland atrophy with dryness of the eyes, mouth, and vagina. The modern concept of Sjögren’s syndrome was firmly rooted in 1933 when Henrik Sjögren, a Swedish ophthalmologist, reported a series of 19 cases of keratoconjunctivitis sicca including two cases with swelling of the major salivary glands.2 Over the next 2 decades, Sjögren and others published extensively about various facets of the disease that now carries his name (also called Gougerot-Sjögren disease), with most of these contributions coming from European ophthalmologists.
In 1953 Morgan and Castleman3 published their detailed histopathologic findings from 18 patients with enlarged lacrimal and salivary glands. This pathologic treatise consisted of cases without apparent etiology that had clinical features resembling those of Mikulicz’s disease and Sjögren’s syndrome. Notably, 15 of the 18 patients were women with lacrimal and salivary gland swelling that developed in the fifth and sixth decades of life. Morgan and Castleman found in the salivary gland tissue a consistent “lymphoid element” accompanied by a striking proliferation of myoepithelial and epithelial cells. The epithelial changes produced a characteristic narrowing or obliteration of the ductal lumen, forming cords of solid cell masses they called epimyoepithelial islands. Because of the prominence of these epithelial structures, they advanced the theory that Mikulicz disease had its origins in the ductal epithelium. Morgan and Castleman also recognized that the salivary gland pathology in their cases was similar to that of the patients described by Henrik Sjögren with keratoconjunctivitis sicca. Because Sjögren’s patients were mostly middle-aged women, Morgan and Castleman reasoned that Mikulicz disease was a subset of Sjögren’s disease but with incomplete clinical manifestations. This now classic paper had the effect of unifying Mikulicz disease and Sjögren’s syndrome into a single disease entity, a belief that dominated the field until recently.
When Joseph J. Bunim delivered the Heberden Oration at the Wellcome Foundation in London on December 2, 1960, he enlightened his audience about the latest advances in Sjögren’s syndrome.4 Bunim described in detail the clinical, pathologic, and laboratory findings of the 40 patients with Sjögren’s syndrome evaluated by himself, Kurt Bloch, Martin Wohl, Richard Oglesby, and Irwin Ship at the Clinical Center of the National Institutes of Health (NIH). All patients in their series had at least two of the three following features: keratoconjunctivitis sicca, xerostomia (with or without enlargement of the salivary glands), and rheumatoid arthritis. Bunim also knew from the work of others that keratoconjunctivitis sicca and xerostomia, or sicca complex, occurred in some patients with systemic lupus erythematosus, scleroderma, polymyositis, and polyarteritis nodosa. He brought home the point that the exocrinopathy extended beyond the lacrimal and salivary glands to affect the pharynx, larynx, and trachea, as well as the vagina. Many of their patients with Sjögren’s syndrome also had extraglandular manifestations such as Raynaud’s phenomenon, purpura, pulmonary infiltrates on chest radiograph, and peripheral neuropathy. Talal and Bunim took note of the diagnosis of reticulum cell sarcoma (an older term that includes non-Hodgkin’s lymphoma) in three patients and Waldenström’s macroglobulinemia in a fourth patient from the NIH cases, drawing attention for the first time to the increased risk of developing lymphoma in this disease.5 In this report, they speculate that a “chronic state of immunologic hyperactivity and the proliferation of immunologically competent cells producing abnormal tissue antibodies predisposes to the relatively frequent development of malignant lymphoma.”5
The NIH group also pioneered the characterization of the serologic markers of Sjögren’s syndrome. In 1965 they reported that 12 of their 16 patients (75%) with sicca symptoms but no evidence of another connective tissue disease had serum antinuclear antibody (ANA) reactivity by indirect immunofluorescence on rat liver tissue.6 Applying the Ouchterlony plate method, they further showed that sera from 13 of 16 (81%) of these patients contained precipitating antibodies to SjD and SjT, which were later to be called anti-Ro (SS-A) and anti-La (SS-B) antibodies. Moreover, Bloch and colleagues7 set the stage for our modern classification schemes by subdividing their patients at the NIH into primary and secondary Sjögren’s syndrome. Secondary Sjögren’s syndrome was the category reserved for patients with sicca symptoms in the setting of another connective tissue disease such as rheumatoid arthritis, systemic lupus erythematosus, scleroderma, or dermatomyositis, whereas primary Sjögren’s syndrome was the designation when the sicca complex occurred in the absence of another connective tissue disease.
In the late 1960s and early 1970s, the investigation of labial salivary gland biopsy for the diagnosis of Sjögren’s syndrome led to the development of grading systems for quantifying the intensity of tissue inflammation. In 1968 Chisholm and Mason proposed a grading system for labial salivary gland biopsies that used a simple grading scale (0 to 4) in which grades 0, 1, and 2 represented absent, slight, and moderate mononuclear cell infiltrates, respectively.8 The higher grades of 3 and 4 corresponded to focal inflammatory cell scores of 1 and greater than 1 per 4 mm2 of tissue, where a focus equated to an aggregate of 50 or more mononuclear cells. In this initial study, 9 of the 10 patients with Sjögren’s syndrome were classified as grade 3 (n = 3) or grade 4 (n = 6). By comparison, biopsies had lower grades when they were taken from patients with other chronic inflammatory conditions, as well as controls. For example, among 10 patients with rheumatoid arthritis, the highest biopsy grade was only 3. Twenty other subjects with a variety of other rheumatic diseases (e.g., osteoarthritis, reactive arthritis, psoriatic arthritis, scleroderma) had biopsies with grades of 0 or 1. Although it was a small study by today’s standards, this work inspired others to validate and refine these findings. Later, postmortem biopsies from 116 controls without a history of an inflammatory disease were scored with only grade 0, 1, or 2 infiltrates, attesting to the diagnostic specificity of this grading system.9 However, the Chisholm and Mason grading system had limitations because it was sensitive only at the lower end of the scale and did not discriminate among biopsies with the highest degree of cellular infiltrates or biopsies with two or more foci per 4 mm2 of tissue.
To improve on these earlier efforts, Tarpley and co-workers10 at the NIH developed a grading system that incorporated scales for not only estimating the extent of inflammation but also quantifying the amount of acinar destruction. As expected, they found that labial salivary gland biopsies from patients with the sicca complex alone had higher grades of cellular infiltrates and acinar destruction than those with sicca symptoms and rheumatoid arthritis or rheumatoid arthritis alone. However, grading the acinar destruction did not add diagnostic value independent of the severity of the infiltrates. Greenspan and colleagues11 from the University of California at San Francisco modified the Chisholm and Mason grading scale when they examined labial salivary gland biopsies from 54 patients with definite or probable Sjögren’s syndrome together with 21 controls. Their results reinforced the diagnostic relevance of the focus score, including its relationship to focus size, and highlighted the presence of germinal centers.11 Grade 4 biopsies (>1 focus per 4 mm2 of tissue) were primarily found in the patients with Sjögren’s syndrome, with the highest focus scores in patients with the sicca complex in the absence of another connective tissue disease. The range for the focus score was extended from 1 to 12, where a score of 12 was arbitrarily assigned to those specimens with foci so numerous that they were confluent. The focus score was thereafter adopted as the “gold standard” for quantifying chronic inflammation in labial salivary gland biopsies. Because the signs and symptoms of xerostomia were relatively nonspecific indicators of salivary gland involvement in Sjögren’s syndrome, other methods such as sialometry, chemical analysis of the saliva, sialography, and scintigraphy were also explored for diagnosing this condition. However, these measures of salivary flow and duct anatomy proved to be diagnostically nonspecific; sialography and scintigraphy had other shortcomings limiting their clinical use.
With a growing interest in Sjögren’s syndrome, investigators began to develop classification criteria for comparison of results across studies. In the 1980s, many groups proposed classification criteria for Sjögren’s syndrome, aiming to identify patients with a sicca complex caused by a chronic autoimmune disorder. These proposed criteria incorporated a combination of the following items: symptoms of the ocular and oral components; objective measures of lacrimal and salivary gland involvement; a focus score greater than or equal to 1 from a labial salivary gland biopsy; and the presence of serum autoantibodies. Many investigators from Europe, the United States, and Japan contributed to this effort, leading to the general acceptance in 2002 of the revised version of the European criteria proposed by the American-European Consensus group.12 The International Sjögren’s Syndrome Registry, funded in 2003, is collecting data that will inform the natural history of Sjögren’s syndrome. It will also support validation of new criteria sets that may be developed from the baseline data of the more than 1200 patients across the world with suspected primary or secondary Sjögren’s syndrome.
Definitions and Classification Criteria
Other terms used to describe Sjögren’s syndrome are autoimmune exocrinopathy13 and autoimmune epithelitis.14 In Sjögren’s syndrome, the term exocrinopathy receives emphasis because of the generalized glandular involvement causing dysfunction of the lacrimal and salivary glands, as well as the apocrine sweat glands of the skin and the submucosal glands of the nose, pharynx, larynx, large airways, and vagina. The term epithelitis gains footing from the uniformly activated epithelial cells omnipresent in the lacrimal and salivary glands and other sites of glandular involvement.
International agreement has been reached on the classification of Sjögren’s syndrome. By the early 1980s, several criteria sets had been proposed for the classification of Sjögren’s syndrome including the Copenhagen criteria,15 the Japanese criteria,16 the Greek criteria,17 and the California criteria.18 Common to each of these criteria sets was the requirement for objective evidence of keratoconjunctivitis sicca and salivary gland involvement. They differed, however, in their item content and weighting, as well as the methods for assessing salivary gland involvement. For example, some of the criteria relied on whole salivary flow (Copenhagen), whereas other criteria relied on parotid flow rate (Greek, California). Only two of the criteria sets discriminated between primary and secondary Sjögren’s syndrome (Copenhagen and Greek).
Movement toward consensus began when the Epidemiology Committee of the European Community conducted a multicenter study involving 26 centers from 12 countries aimed at developing criteria for the classification of Sjögren’s syndrome. This group first agreed on the items that should be included in the classification criteria for Sjögren’s syndrome and then tested their operational characteristics in a large sample of patients with a clinical diagnosis of primary Sjögren’s syndrome (n = 246), secondary Sjögren’s syndrome (n = 201), other connective tissues diseases without Sjögren’s syndrome (n = 113), and healthy controls (n = 133).19 Two sets of three questions were selected from a larger body of questions that best correlated with the presence of keratoconjunctivitis sicca and xerostomia, respectively, as judged by clinical experts. The results from objective tests were analyzed by univariate analyses, yielding a tentative list of items based on their sensitivity and specificity for correct classification of the diagnosis. The following six items were chosen for the classification criteria: (I) ocular symptoms; (II) oral symptoms; (III) ocular signs (Schirmer-I-test ≤5 mm/5 min or Rose Bengal score ≥4 by the van Bijsterveld scoring system); (IV) histopathologic features (focus score ≥1 on labial salivary gland biopsy; (V) objective evidence of salivary gland involvement by at least one abnormal test (salivary scintigraphy, parotid sialography, or unstimulated salivary flow rate ≤1.5 mL/15 min); and (VI) at least one of the following serum autoantibodies: anti-Ro/SS-A or anti-La/SS-B antibodies, ANAs, or rheumatoid factor. Exclusion criteria were pre-existing lymphoma, acquired immunodeficiency syndrome, sarcoidosis, and graft-versus-host disease. The presence of four of six criteria (accepting only a positive test for anti-Ro/SS-A or anti-La/SS-B antibodies for item VI had good sensitivity (93.5%) and specificity (94%) for correctly classifying a patient with primary Sjögren’s syndrome. It was suggested to be the optimal combination for efficient classification of this condition. For classification of secondary Sjögren’s syndrome, the best combination was found to be a positive response for items I or II plus a positive result for any two of items II, IV, and V, resulting in a sensitivity of 85.1% and specificity of 93.9% for this diagnosis. With a positive response to items I or II and only one positive response to items III to V, the sensitivity increased to 95.6% but the specificity dropped to 71.6%.
These preliminary European classification criteria for Sjögren’s syndrome were validated in a second study of similar design. Cases of primary Sjögren’s syndrome (n = 81), secondary Sjögren’s syndrome (n = 76), other connective tissue diseases without Sjögren’s syndrome (n = 54), and controls (n = 67) were assembled by expert clinicians at 16 centers from 10 countries to validate the initial results.20 In this study, it was decided not to use items III (a) (Schirmer-I-test) and V (c) (unstimulated whole salivary flow) to classify patients older than age 60 because the tear and salivary flow by these measures were significantly reduced in the elderly controls from the original study. The results proved to be similar to those of the earlier study, showing a sensitivity of 97.5% and specificity of 94.2% for correctly classifying primary Sjögren’s syndrome and a sensitivity of 97.3% and specificity of 91.8% for correctly classifying secondary Sjögren’s syndrome. A limitation of this type of study design is the possibility that the sensitivity and specificity of these criteria may be artificially inflated by a “circular bias” deriving from the results of the diagnostic testing on the original selection of patients. These preliminary European classification criteria have also been criticized because a patient may be classified with primary Sjögren’s syndrome despite a negative biopsy and the absence of serum anti-Ro/SS-A or anti-La/SS-B antibodies. Thus patients may be classified as having primary Sjögren’s syndrome without evidence of an immune basis for their condition.
This conceptual impasse was overcome by the European Study Group on Classification Criteria for Sjögren’s Syndrome with a group of American experts. From their previous study, they selected a cohort of 76 patients with primary Sjögren’s syndrome, 41 patients with another connective tissue disease but without Sjögren’s syndrome, and 63 controls and tested three different combinations of items from the original classification criteria using a receiving operator curve (ROC) analysis. To determine the best possible prediction model, the optimal classifiers were compared with ROC analysis for the three different combinations: C point (positivity for any four of the six items); C* point (positivity of any four of the six items, excluding cases negative for both items IV and VI); and D point (positive for any of the four objective criteria).12 The C point and the C* point showed the same accuracy (92.7%); however, the C* point had a lower sensitivity than the C point (89.5% vs. 97.4%), but a higher specificity (95.2% vs. 89.4%), and was preferred on the basis of the goal of selecting criteria with a high probability of excluding patients without the disease. The sensitivity and specificity of the D point were 84.2% and 95.2%, respectively, and judged to be comparable with the C* point and acceptable for classification purposes. Therefore patients may now be classified as having primary Sjögren’s syndrome if they fulfill four of the six items including either a positive labial salivary gland biopsy or a positive test for serum anti-Ro/SS-A and/or anti-La/SS-B antibodies. This combination avoids the problem of classifying a patient with primary Sjögren’s syndrome in the absence of a positive labial salivary gland biopsy or a positive test for anti-Ro/SS-A and/or anti-La/SS-B antibodies. Alternatively, they may meet the criteria by satisfying any four of the objective items, which was a highly unusual scenario in their data set because nearly all patients with primary Sjögren’s syndrome manifested symptoms of dry eyes and/or dry mouth.
The revised classification criteria proposed by the American-European Consensus Group are shown in Table 73-1. It was further specified that the Schirmer-I test be performed with anesthesia and that other ocular dyes such as lissamine green (for conjunctival staining) and fluorescein (for corneal staining) be allowed as replacements for Rose Bengal dye, which was not available in many countries. A positive labial salivary gland biopsy was further defined according to the rules set forth by Daniels and Whitcher.21 In this case, a positive biopsy must show evidence of focal lymphocytic sialadenitis (FLS), which they defined as dense aggregates of 50 or more lymphocytes in perivascular or periductal locations. These aggregates, or foci, must contain only a small proportion of plasma cells and be located adjacent to normal-appearing acini in lobules without duct dilatation or fibrosis. A minimum threshold for positivity was considered to be greater than or equal to 1 focus per 4 mm2 of tissue. These rules are important to follow in the interpretation of labial salivary gland biopsies because many specimens, especially from elderly individuals, show patterns of inflammation consistent with chronic sialadenitis, namely mixed lymphocytic and plasma cell infiltrates in association with ductal dilation, acinar atrophy, and fibrosis.
Table 73-1 Revised International Classification Criteria for Sjögren’s Syndrome (SS)
Reproduced from Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group, Vitali C, Bombardieri S, Jonsson R, et al and the European Study Group on Classification Criteria for Sjögren’s Syndrome, Ann Rheum Dis 61:554–558, 2002 with permission from BMJ Publishing Group Ltd.
An abnormal parotid sialogram has been clarified in the revised criteria as the presence of diffuse sialectasis according to the scoring system of Rubin and Holt.22 Positivity by salivary scintigraphy was also further defined as delayed uptake, reduced concentration, or delayed secretion of the tracer, using the method of Schall and colleagues.23 Parotid sialography and salivary scintigraphy are rarely used clinically in the United States. The revised version also included some modifications to the list of exclusion criteria.
These new revised criteria have also not escaped criticism by some experts because of the possibility that patients may be classified as having primary Sjögren’s syndrome without subjective or objective evidence of ocular involvement (satisfying items IV, V, and VI).24 Others have mentioned the “histologic and immunologic bias” of the revised criteria,25 but this argument fails if the goal of the classification criteria is to define patients with an immune-mediated condition. Some patients with primary Sjögren’s syndrome will test positive for serum ANAs in the absence of anti-Ro/SS-A and anti-La/SS-B antibodies. If such patients have a falsely negative labial salivary gland biopsy, then they will not be classified as having primary Sjögren’s syndrome despite a better than average chance otherwise. Another potential shortcoming is the lack of laboratory standardization for the detection of anti-Ro/SS-A and anti-La/SS-B antibodies. Many different methodologies are now available to measure these autoantibodies including the older Ouchterlony plate assay, various commercial enzyme-linked immunosorbent assay (ELISA) kits, and the newer multiplex bead technology. It is unclear if the type of methodology influences the sensitivity or specificity of these criteria. Although these classification criteria have been generally accepted by the medical community, they have not been officially endorsed by the American College of Rheumatology or the European League Against Rheumatism.
Recently, new classification criteria have been published based on data from the International Collaborative Clinical Alliance Cohort.25a These criteria have been approved by the American College of Rheumatology. These criteria are derived solely from objective test results and require for classification of primary Sjogren’s syndrome at least 2 of the following: (1) a positive test for serum anti-Ro/SS-A and/or anti-La/SS-B antibodies or a positive test for rheumatoid factor and an ANA titer of ≥1 : 320; (2) labial salivary gland biopsy exhibiting focal lymphocytic sialadenitis with a focus score ≥44 mm2; and (3) keratoconjunctivitis sicca with ocular staining score ≥3. Using an external set of 303 participants, these classification criteria had a sensitivity of 92.5% and a specificity of 95.4%. Further validation of these criteria will be necessary to confirm their performance.
Epidemiology
Primary Sjögren’s syndrome ranks among the most common of the autoimmune diseases, with a prevalence rate ranging from 0.1% to 4.6%.26 However, the epidemiologic data are confounded by variations in the ages of the study populations and differences in the classification criteria used for case identification. For example, the application of the Copenhagen criteria yields a prevalence rate for Sjögren’s syndrome 2.7-fold greater than the California criteria, whereas the preliminary European classification criteria captures 2- to 3-fold more patients than the Copenhagen criteria.27 Bowman and colleagues28 estimated the prevalence of primary Sjögren’s syndrome at 0.1% to 0.6% in a community from the United Kingdom using the revised criteria proposed by the American-European Consensus Group. In a study from Greece employing these same criteria, the age-adjusted mean annual incidence and prevalence rates were 5.3 (confidence interval [CI], 4.5 to 6.1) per 105 persons (0.5 for men and 10.1 for women) and 92.8 per 100,000 persons (8.4 for men and 177.4 for women), respectively.29 The incidence and prevalence rates of primary Sjögren’s syndrome are significantly higher in women than men (e.g., approximately 20 : 1), with a peak incidence in the fifth and sixth decades of life.29 Primary Sjögren’s syndrome occurs infrequently in children with onset as early as 5 years of age.30 The revised criteria of the American-European Consensus Group may be less sensitive for classifying primary Sjögren’s syndrome in children than adults owing to differences between these two age groups in the clinical presentation of this disease.30
The prevalence of secondary Sjögren’s syndrome associated with rheumatoid arthritis, systemic lupus erythematosus, and systemic sclerosis has been estimated to be 17.1%,31 8% to 20%,32–34 and 14%35; respectively. To be classified as secondary Sjögren’s syndrome, patients in these studies had at minimum symptoms of keratoconjunctivitis sicca or xerostomia and objective evidence of lacrimal or salivary gland involvement. None of these studies used the preliminary European classification criteria for secondary Sjögren’s syndrome.
Genetics and Pathogenesis
Because a disproportionately high number of the cases of primary Sjögren’s syndrome occur in women, the search for environmental candidates has centered on abnormal regulation of estrogens and androgens. However, no major differences have been found between patients with primary Sjögren’s syndrome and healthy controls in serum levels of sex steroid hormones.36 In addition, treatment with the hormone dehydroepiandrosterone, which acts on the androgen receptor, has shown no clinical efficacy in women with primary Sjögren’s syndrome.37 Among possible viral triggers, Epstein-Barr virus (EBV) and cytomegalovirus (CMV) have received attention in primary Sjögren’s syndrome due to their suppressive effects on T cell immunity and their ability to establish persistent infection. In one study, EBV deoxyribonucleic acid (DNA) was found by immunochemical staining in salivary gland biopsies to localize in acinar and ductal epithelial cells38; however, the available evidence does not otherwise support a direct role for EBV infection in the pathogenesis of this disease. A 94-bp fragment of cocksackievirus ribonucleic acid (RNA) was also shown to be differentially expressed in salivary gland biopsies from patients with primary Sjögren’s syndrome compared with controls,39 but these results were not replicated in a later study.40 As yet, the identity of a possible viral trigger remains elusive, if indeed one exists.
It has been difficult to estimate the genetic risk for developing primary Sjögren’s syndrome owing to the absence of large twin studies. However, the existence of many large families with two or more members with primary Sjögren’s syndrome argues strongly for a genetic component to disease pathogenesis.41 Primary Sjögren’s syndrome is considered to be a complex genetic disorder, similar to the genetic susceptibility of systemic lupus erythematosus and rheumatoid arthritis. It is now clear from genetic studies of human autoimmune diseases that multiple genes contribute to disease risk and that individually each gene confers only modest effects on disease susceptibility.42 The exception to this rule is the relatively strong signal associated with the human leukocyte antigen (HLA) locus on human chromosome 6p21.3. In populations of European descent, confirmed HLA associations with primary Sjögren’s syndrome include DRB1*0301 (DR3), DRB1*1501 (DR2), DQA1*0103, DQA1*0501, DQB1*0201, and DQB1*0601.43,44 The disease-associated polymorphisms located in the DRB1*0301 and DRB1*1501 loci account for 90% of the HLA genetic contribution. The HLA locus appears to play a major role in the pathogenesis of autoantibody responses associated with primary Sjögren’s syndrome. In patients with primary Sjögren’s syndrome, higher titers of anti-Ro/SS-A and anti-La/SS-B antibodies have been linked to heterozygosity for the DQA1 and DQB1 alleles.45
The genetic susceptibility to primary Sjögren’s syndrome will likely include inheritance of genes identified as risk factors in other autoimmune diseases. Familial clustering of cases of primary Sjögren’s syndrome with systemic lupus erythematosus, rheumatoid arthritis, and systemic sclerosis, as well as other autoimmune diseases, are consistent with this premise.46–48 There have been no large-scale genome-wide association studies in primary Sjögren’s syndrome. Most efforts to identify disease susceptibility genes in primary Sjögren’s syndrome have taken the candidate gene approach. Genes in the type I interferon (IFN) pathway have been the focus of several candidate gene studies because they are highly expressed in the peripheral blood and salivary glands of patients with primary Sjögren’s syndrome compared with controls.49 In a small cohort study, an increased risk of primary Sjögren’s syndrome was associated with a single nucleotide polymorphism in the splicing sequence of exon B of the gene encoding IFN regulatory factor 5 (IRF5).50 IRF5 is among the nine IRFs that signal through Toll-like receptors (TLRs) and is essential for inducing responses through TLR4, 7, and 9.51 Given the possible inciting roles of viral RNA and DNA, the endosomal TLR7 and TLR9 pathways may be important in primary Sjögren’s syndrome for the activation of the type I IFN pathway.52 A 5-bp insertion/deletion polymorphism (CGGGG insertion/deletion) in the promoter region of the IRF5 transcript has also been linked to primary Sjögren’s syndrome.53 This polymorphism appears to have functional significance because reovirus (double-stranded RNA virus)–infected salivary gland epithelial cells in culture bearing this polymorphism produce higher levels of the IRF5 transcript.53
Other susceptibility genes have been identified in primary Sjögren’s syndrome, but the findings have not yet been widely replicated. In a case-control study, an increased risk for primary Sjögren’s syndrome was associated with a variant haplotype of the transcription factor Signal Transducers and Activator of Transcription 4 (STAT4).54 This STAT4 polymorphism has also been shown to increase the risk for systemic lupus erythematosus and rheumatoid arthritis. STAT4 is a key intracellular signaling molecule involved in IL-12 and IL-23 signaling and is known to promote the development of T helper 1 (Th1) and T helper 17 (Th17) responses. Polymorphisms in the STAT4 and IRF5 genes appear to be additive in the risk for developing primary Sjögren’s syndrome.55 In other studies, Nordmark and colleagues56 have identified three gene loci associated with an increased risk of primary Sjögren’s syndrome—the early B cell factor (EBF1) gene, the interval encompassing family with sequence similarity 167 member A and B lymphoid tyrosine kinase (FAM167A-BLK), and the tumor necrosis family member 4 (TNFSF4, or the OX40L gene). All three genes are involved in B cell development, which is of particular interest due to the hyperactivated state of B cells in this disease.
The human genome contains conserved noncoding elements with functional sequences including regulatory motifs in promoters and untranslated regions of genes. Some of these conserved noncoding elements encode microRNAs, a novel means for regulation of gene expression. MicroRNAs influence both innate and adaptive immunity and have been shown to play a role in late B cell differentiation and development, as well as the establishment of B cell tolerance. Early studies have shown that microRNA expression patterns in salivary gland tissue can distinguish patients with Sjögren’s syndrome from controls, suggesting a pathologic role for dysregulated microRNA expression in the regulation of the chronic inflammatory response.57
Many insights have been gained about disease mechanisms in primary Sjögren’s syndrome from studies of humans with this autoimmune disease, as well as animal models. Experiments in model systems enable hypotheses to be examined in ways not possible in humans and have contributed to understanding the immunoregulatory disturbances underlying the clinical expression of disease (Table 73-2).58,59 Whereas these animal models have shed light on disease mechanisms, it has been possible to perform highly significant research in humans with primary Sjögren’s syndrome because of the accessibility of labial salivary gland tissue for immunohistopathologic analysis. Studies of labial salivary gland biopsies from patients with this disease have shown approximately 90% of the infiltrating cells are composed of CD4+ T lymphocytes and B lymphocytes, with the remainder an admixture of plasma cells, CD8+ T cells, FoxP3+ T regulatory cells, CD56+ natural killer (NK) cells, and macrophages, as well as myeloid and plasmacytoid dendritic cells (DCs)60 (Figure 73-1). Most of the infiltrating T cells bear the memory phenotype (CD45RO) and display a restricted T cell receptor (TCR) repertoire representing several different clonotypes across multiple Vβ families. The proportion of B cells in the infiltrate increases with the severity of the inflammatory lesion.
Table 73-2 Mouse Models of Sjögren’s Syndrome*
| Mouse Model | Phenotype | Comments |
|---|---|---|
| Spontaneous Disease Models | ||
| (NZB)NZW F1 mice | Progressive focal sialadenitis | Glandular involvement F > M |
| MRL/lpr | Lymphocytic infiltration of lacrimal and salivary glands; anti-Ro/SS-A and MR3 antibodies; oligoclonal expansion of T cells and IgA and IgM production in the salivary glands | Normal secretory function; mRNAs for IL-1 and TNF expressed in salivary glands before onset of sialadenitis |
| NOD and its derivatives NOD.H2h4, NOD.Q, and NOD.P, NOD.E2fl−/−, NOD.scid | Lymphocytic infiltration of lacrimal and salivary glands; NOD.H2h4 strain (but not parental NOD strain) secretes high levels of anti-Ro/SS-A and anti-La/SS-B antibodies; reduced glandular function | Mice also develop diabetes; exchange of H2 haplotype from H2g7 to H2q (NOD.Q) or H2p (NOD.P) does not affect the frequency of sialadenitis; disruption of ICA69 locus prevents lacrimal gland inflammation and reduces salivary gland inflammation; NOD IFN-γ−/− mice do not develop glandular disease; blockade of LT-βR signaling pathway reduces salivary gland infiltrates and improves salivary gland function |
| NFS/sld | Lymphocytic infiltration of lacrimal and salivary glands; anti-α-fodrin antibodies | Aberrant immune responses to α-fodrin; mice develop autoimmune lesions in other organ systems |
| Experimentally Induced Models | ||
| Carbonic anhydrase (PL/J mice) | Lymphocytic infiltration of salivary glands; antibodies to carbonic anhydrase | |
| Ro peptides (Balb/c mice) | Lymphocytic infiltration of salivary glands; glandular hypofunction, anti-Ro/SS-A and anti-La/SS-B antibodies | Disease induction requires multiple injections of peptide emulsified in Freund’s adjuvant |
| Transgenic or Knockout Models | ||
| Id3−/− | Lymphocytic infiltration of lacrimal and salivary glands; adoptive transfer experiments; anti-Ro/SS-A and anti-La/SS-B antibodies; salivary hyposecretion | Id3 gene involved in TCR-mediated T cell indicate a role for T cells in the development of disease; treatment with anti-CD20 antibodies ameliorates disease |
| PI3K−/− | Lymphocytic infiltration of the lacrimal glands; anti-Ro/SS-A and anti-La/SS-B antibodies | |
| BAFF transgenic | Lymphocytic infiltration of lacrimal and salivary glands; unique population of marginal zone B cells in salivary glands | No anti-Ro/SS-A or anti-SS-B antibodies; also develop lupus manifestations and anti-DNA antibodies and RF |
| IL-14α transgenic | Lymphocytic infiltration of lacrimal and salivary glands; hypergammaglobulinemia; <25% of animals develop anti-Ro/SS-A and anti-La/SS-B antibodies; glandular hyposecretion; mild immune complex-mediated renal disease and lymphocytic interstitial pneumonitis | IL-14 is a growth factor for B cells; mice develop large B cell lymphomas later in life; role for LT-α in the salivary gland inflammation; infiltrate primarily B cells with relatively few CD4+ and CD8+ T cells |
| IL-12 transgenic | Lymphocytic infiltration of lacrimal and salivary glands; anti-La/SS-B antibodies; glandular hyposecretion; increased acinar cell volume | Mice also develop thyroiditis and lung pathology |
BAFF, B cell activating factor; F, female; ICA69, islet cell antigen 69; Id3, protein inhibitor of DNA binding 3; IFN-γ, interferon-γ; IL-1, interleukin-1; LT-α, lymphotoxin-α; LT-βR, lymphotoxin β receptor; M, male; MR3, muscarinic receptor subtype 3; NOD, nonobese diabetic; NZB/NZW F1, New Zealand Black × New Zealand White F1 (mouse hybrid); PIK3, phosphoinositide 3-kinase; TNF, tumor necrosis factor.
* See references 58 and 59 for further details.
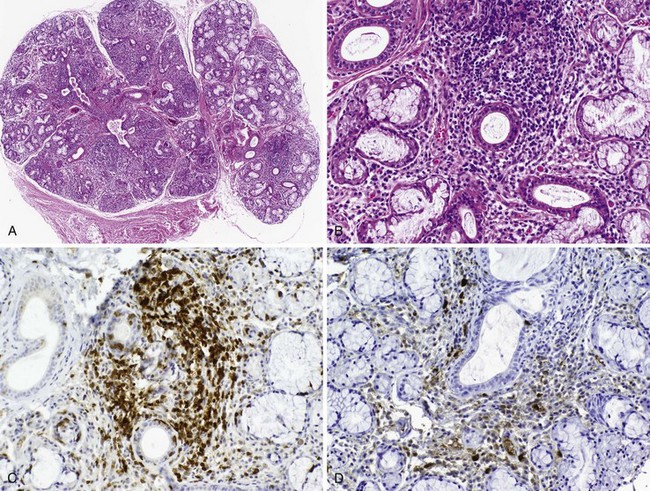
The infiltrating mononuclear cells tend to coalesce around ducts and blood vessels, and in more severe inflammatory lesions they may form aggregates organized into germinal center (GC)-like structures. The GC-like structures display well-circumscribed mononuclear cell infiltrates with B and T cell components, Ki-67+ proliferating cells, CD21/CD35+ follicular DC networks, and CD31+ high endothelial venules (HEVs).61 CXCL13 expression by epithelial cells, HEVs, and within germinal center–like structures together with the expression of CXCL12 and CCL21 provides a salivary gland microenvironment capable of attracting and retaining B cells.62–64 The myeloid DCs and macrophages, the classic antigen-presenting cells, are mostly found in proximity to the ductal epithelium, where they have been shown to secrete tumor necrosis factor (TNF), interleukin-6 (IL-6), IL-10, IL-12, and IL-18. Minor salivary glands also contain a small number of plasmacytoid DCs, the main producers of the type I IFNs.65 A robust type 1 IFN signature has been detected both in the salivary glands and peripheral blood of patients with primary Sjögren’s syndrome.65,66 The generation of type 1 IFNs by plasmacytoid DCs, among the first lines of defense against viral infection, is partially dependent on signals through TLR7 and TLR9.
The analysis of T cell cytokines in labial salivary gland biopsies from patients with primary Sjögren’s syndrome suggests a predominantly Th1- and Th17-driven response.67 Studies of the expression of cytokine messenger RNAs (mRNAs) in salivary gland tissue show mainly upregulation of IL-2 and IFN-γ, the Th1-specific cytokines, and lesser quantities of IL-4, IL-5, and IL-13, the Th2-specific cytokines. Th17 cells, so named because they produce IL-17, are also found in the minor salivary glands; the salivary gland microenvironment is also rich in transforming growth factor (TGF)-β, IL-6, and IL-23, which are cytokines known to promote the development of this subset.68 In addition, the salivary gland infiltrates contain a generous infiltrate of FoxP3+ regulatory T cells.69 These cells typically show suppressive behavior, but as yet they have an uncertain role in regulating the chronic inflammatory lesion.
Several features of primary Sjögren’s syndrome implicate B cells in disease mechanisms. B cells are the source of autoantibodies, but they also activate T cells through their ability to present antigen, are stimulated to secrete proinflammatory and anti-inflammatory cytokines, and facilitate the organization of secondary and tertiary lymphoid tissue. The frequent presence of hypergammaglobulinemia, circulating immune complexes, mixed monoclonal immunoglobulin M (IgM) cryoglobulinemia, and serum autoantibodies in patients with primary Sjögren’s syndrome implies B cells are in a dysregulated state. Most B cells in the salivary gland tissue exhibit a memory phenotype, express somatically hypermutated immunoglobulin V genes, and show preferential changes in VL gene usage and the length of CDR3 from VH gene rearrangements, which are findings characteristic of an antigen-driven response.70 Salivary gland B cells appear to be responding specifically to antigen, although the site of antigen encounter and activation (salivary gland vs. secondary lymphoid tissue) remains uncertain. Germinal center–like structures found in some of the salivary gland biopsies provide an opportune environment for the selection and differentiation of antigen-driven B cell responses. The distribution of peripheral blood B cell subsets in primary Sjögren’s syndrome differs from healthy controls, showing increased proportions of CD27− (naïve) B cells and decreased proportions of CD27+ (memory) B cells.71 Among several possibilities, these changes in the peripheral blood may result from abnormal trafficking of CD27+ memory B cells to the target tissues, aberrations in B cell development, or both.
Several intrinsic B cell defects have also been found in patients with primary Sjögren’s syndrome including abnormal retention of preswitch Ig transcripts in circulating postswitch CD27+ memory B cells72 and enhanced B cell signaling due to altered kinetics of B cell receptor translocation to lipid rafts.73 Moreover, studies in patients with primary Sjögren’s syndrome have shown increased serum and salivary gland tissue levels of B cell activating factor (BAFF), a B cell prosurvival factor,74 as well as upregulated salivary gland expression of lymphotoxin (LT)-β mRNA.75 LT-β is required for the formation of lymph nodes and germinal centers, while the heterodimer LT-α/LT-β can induce the development of ectopic germinal center–like structures. LT-α in soluble form induces the secretion of IFNs and chemokines, which are elevated in the salivary gland tissue from patients with primary Sjögren’s syndrome. Levels of IL-14, another B cell growth factor, also appear to be increased in the serum and saliva of patients with primary Sjögren’s syndrome.76 IL-14 transgenic mice have been shown to develop a Sjögren’s-like phenotype77 in which the local tissue response is critically dependent on LT-α.78 Blocking LT-β receptor signaling in NOD mice ablates the lymphoid organization in the salivary glands and improves their function,79 further evidence that LTs may play a role in the pathogenesis of Sjögren’s syndrome. Finally, abnormal B cell behavior is implied in primary Sjögren’s syndrome by the predisposition of this disease toward the development of non-Hodgkin’s B cell lymphoma. Interestingly, IL-14 transgenic mice develop large B cell lymphomas later in life.77
Anti-Ro/SS-A and anti-La/SS-B antibodies do not appear to have a pathogenic role in disease mechanisms despite their diagnostic and prognostic significance. The stimulating antigen(s) is unknown. An aberrant response to self may be provoked by altered expression of autoantigens. Several models have been proposed to explain the altered expression of autoantigens including differential expression of protein isoforms, post-translational modification, and abnormal autoantigen presentation via apoptotic blebs, exosomes, or heat shock protein–mediated cross-priming.80 Ro/SS-A and La/SS-B proteins do in fact show upregulated expression in the vicinity of the immunopathologic lesion in primary Sjögren’s syndrome,80 where they may induce local immune responses. Saliva from patients with this disease has been shown to contain anti-Ro/SS-A and anti-La/SS-B antibodies.81 Although this finding may be interpreted to reflect local production of autoantibodies, it may also reflect extravasation of proteins from blood vessels into the inflamed tissues.
Among the other autoantibody specificities, those directed against the muscarinic receptor (MR) have attracted the most interest because of their possible role in causing glandular hypofunction. The MR family of receptors is bound by acetylcholine, which mediates the effects of the preganglionic and postganglionic parasympathetic nerve fibers that primarily regulate salivary flow. The muscarinic type 3 receptor (M3R), the target of autoantibodies in primary Sjögren’s syndrome, is the subtype that predominately controls salivary flow. Two lines of evidence support the hypothesis that anti-M3R antibodies reduce exocrine gland function. Firstly, serum immunoglobulins from patients with primary Sjögren’s syndrome have been shown to bind MR3 receptors on acinar cell membranes; they also have been shown to inhibit acetylcholine-evoked Ca2+ responses in a salivary gland cell line.82 This Ca2+-sensitive response is tightly regulated through intracellular signaling pathways that open the Cl− channels on the apical membrane, resulting in an osmotic gradient and movement of water into the duct lumen. Secondly, passive transfer of the antibody has been shown to cause glandular hypofunction in NOD mice, an animal model of Sjögren’s syndrome.83 However, the assays for detecting serum anti-M3R antibodies have produced variable results depending on the methodology. For this reason, it has been difficult to determine the diagnostic sensitivity and specificity of serum anti-M3R antibodies in primary Sjögren’s syndrome and the possible relationship of these autoantibodies to glandular hypofunction. Other autoantigens have been identified in patients with primary Sjögren’s syndrome including α-fodrin, poly(ADP)ribose polymerase, carbonic anhydrase, and ICA69 protein. Despite initial enthusiasm for the possible etiologic role of α-fodrin in primary Sjögren’s syndrome, recent studies have shown that serum anti-α-fodrin antibodies are not highly specific for this diagnosis.84 Autoantibodies to carbonic anhydrase, poly(ADP)ribose polymerase, and ICA69 protein occur in only a minority of patients with primary Sjögren’s syndrome and therefore do not appear to be attractive candidates for the initiating autoantigens.
In primary Sjögren’s syndrome, the glandular epithelial cell appears to be an active participant in the abnormal immune response. In the salivary gland, the mononuclear cells preferentially congregate around ductal epithelium. The epithelial cells show upregulated expression of HLA class I and II molecules; adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1; CD54), vascular cell adhesion molecule-1 (VCAM-1; CD106) and E-selectin; and co-stimulatory molecules such as CD80 and CD8685; they also produce high levels of cytokine mRNAs for IL-1, IL-6, IL-12, IL-18, and TNF,86–88 as well as BAFF.74 Ductal epithelial cells have also been shown to express mRNAs for several different proinflammatory chemokines including CXCL13, CCL17, CCL21, and CCL22.89 Cultured salivary gland epithelial cells also express functional TLR3 and TLR7 molecules,90,91 endowing a capability to sense pathogens and endogenous molecules produced by injured tissues. Activated epithelial cells are likely driving the aberrant innate and adaptive immune responses not only in the lacrimal and salivary glands but also in the lungs and kidneys, as well as other extraglandular sites.
At least two models exist to explain the glandular hypofunction in Sjögren’s syndrome. In one model, it may be hypothesized that the glandular tissue is destroyed by an immune onslaught perpetuated by persistent exposure to self-antigens or other environmental stimulants (e.g., viral infection), leading to apoptosis of acinar epithelial cells and the irreversible loss of salivary gland function92 (Figure 73-2A). A weakness of this model is that studies suggest epithelial cells rarely undergo apoptosis in the salivary gland tissue despite the fact they upregulate expression of mediators of cell death such as Fas, Fas ligand, and Bax (B cell lymphoma 2–associated X protein).93 Ligand binding to CD40 expressed on epithelial cells in the salivary gland tissue also leads to Fas-mediated cell death by downregulating c-FLIP (cellular FLICE-like inhibitor protein), an inhibitor of Fas-mediated cell death.94 Because no studies have yet shown that an imbalance truly exists between the rates of glandular proliferation and damage, the relevance of this model to glandular hypofunction is uncertain. It also does not explain why many patients with markedly diminished salivary flow retain substantial amounts of normal-appearing acinar tissue in their salivary glands.95
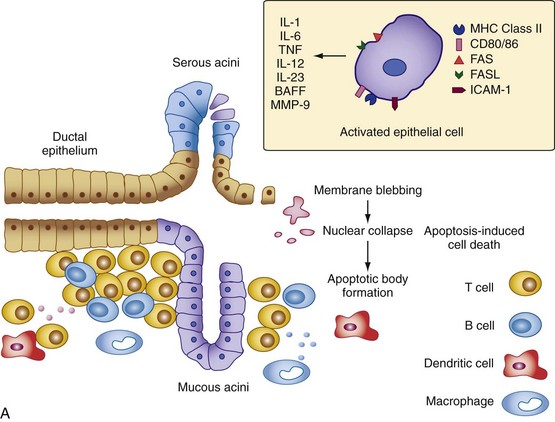
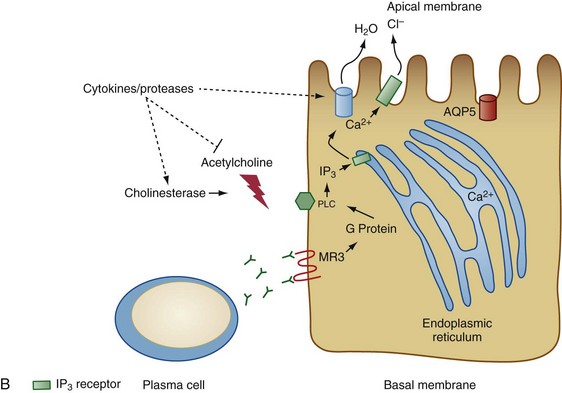
Figure 73-2 Two models for the pathogenesis of salivary gland inflammation in Sjögren’s syndrome. A, Glandular hypofunction is explained by tissue loss secondary to immune attack, resulting in cytotoxic cell death and apoptosis. Epithelial cells likely play a central role in this process by several mechanisms: antigen-presentation and T cell activation; production of proinflammatory cytokines such as interleukin-1 (IL-1), IL-6, tumor necrosis factor (TNF), IL-12, and IL-23; and secretion of proteases. Epithelial cells also upregulate expression of Fas (FAS) and Fas ligand (FASL), cell surface molecules involved in the activation of apoptotic pathways. Other immune cells such as T cells, B cells, macrophages, and dendritic cells serve to amplify the chronic inflammatory response. In model B, glandular hypofunction results from downregulation of receptor-mediated secretion of salivary fluid into the ductal lumen. Acetylcholine binds to muscarinic receptor type 3 (MR3) on the surface of acinar cells, stimulating production of the second messenger inositol 1,4,5-triphosphate (IP3). IP3 in turn diffuses through the cytoplasm until it binds its receptor, IP3R, on the endoplasmic reticulum. This interaction causes calcium to be released into the cytoplasm, which opens Ca2+-sensitive chloride channels on the apical membrane of the cell. Electrochemical neutrality is preserved when Na+ follows chloride across the membrane, while the osmotic gradient propels the water into the ductal lumen. Mechanisms of immune-mediated glandular hypofunction might include inhibition of acetylcholine release by cytokines, increased breakdown of acetylcholine in the epilemmal space by upregulated production of cholinesterases, blockade of M3R by autoantibodies, inhibition of intracellular signaling pathways involved in the fluid secretory process, or altered expression of aquaporin 5 (AQP5), which appears to be primarily responsible for water movement through the apical cell membrane. See reference 96 for more details. BAFF, B cell activating factor; ICAM-1, intercellular adhesion molecule-1; MHC, major histocompatibility complex; MMP-9, matrix metalloproteinase-9; PLC, phospholipase C.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


