117 Sarcoidosis
Sarcoidosis occurs worldwide and affects people of all racial and ethnic backgrounds.
Rheumatologic manifestations are common in sarcoidosis and are often overlooked or misdiagnosed.

 Supplemental image available on the Expert Consult Premium Edition website.
Supplemental image available on the Expert Consult Premium Edition website.
Sarcoidosis is an orphan, systemic, clinically heterogeneous disorder. Its cause has yet to be identified, but environmental, genetic, and infectious causes have been suggested. The hallmark of sarcoidosis is the development and accumulation of noncaseating granulomas in any organ system. Organ system involvement, which is unpredictable and varies between patients, is the major determinant of morbidity and mortality in sarcoidosis. Although any organ system can be involved, the lungs are affected in most cases. Lymphatic, skin, and ocular findings are also common. Given the variability of sarcoidosis manifestations, diagnosing this disorder is often difficult. Patients may be asymptomatic or may present with a range of nonspecific symptoms, but specific symptoms such as cough, dyspnea, burning of eyes, or rash may suggest the diagnosis.1 When extrapulmonary symptoms develop, they sometimes result in rheumatologic manifestations including but not limited to arthritis, skin lesions, arthralgias, and neuropathy.2
Epidemiology
Sarcoidosis is a global disease. Because of its clinical heterogeneity and its variable diagnostic criteria in different countries, the worldwide prevalence and incidence of sarcoidosis have been difficult to calculate. In Northern Europe, up to 40 cases per 100,000 people have been reported.3,4 A study from Eastern Europe found only 3.68 cases of sarcoidosis per 100,000 people.5 The incidence of sarcoidosis in Japan is also low, with one study estimating it at 3.7 cases per 100,000 individuals.6 Within countries, incidence rates may vary between races. In the United States, the annual incidence of sarcoidosis is more than three times higher in black individuals (35.5 per 100,000) than in white people (10.9 per 100,000).7 Furthermore, the disease course of sarcoidosis is more progressive and may be more fatal in black Americans.8,9 Despite numerous epidemiologic studies of sarcoidosis, many clinicians and researchers believe that estimates of prevalence and incidence are lower than actual rates of the disease owing to inaccurate diagnoses or asymptomatic cases that are never diagnosed.
Although sarcoidosis affects men and women of all ages and from diverse ethnic backgrounds,4,10 some disparities in how it affects these groups have been noted. The fact that it affects slightly more women than men has been confirmed in studies from around the world; estimates indicate that 57% of patients with sarcoidosis are women.4 Relative to men, women with sarcoidosis have a greater number of ocular and neurologic manifestations. People of any age may acquire the disease, but the median age of onset is around 40.4 A second peak of incidence has been reported around age 65, especially in women.8,11
Immunopathogenesis
The pathogenesis of sarcoidosis likely involves the interplay of many different cells, cytokines, and other inflammatory mediators. Granuloma development is the characteristic pathologic feature of tissue involvement in sarcoidosis. Physiologically, granulomas act as shields, protecting tissues from pathogens, thereby pre-empting inflammatory reactions. Their formation is the end product of a coordinated effort involving T cell activation, antigen-presenting cell (APC) activation, and cell signaling. Granulomas consist of a core of mononuclear phagocytes, such as epithelioid cells, multinucleated giant cells, and macrophages, which are encased by lymphocytes, including B cells, CD4+ T cells, and CD8+ T cells.12
Innate Immunity
T cell activation is required for granulomas to form. Early in the course of sarcoidosis, an unknown antigen or multiple antigens activate T cells and macrophages, thereby triggering downstream signaling from both cell types. Locally activated CD4+ T helper cells differentiate into T helper type 1 (Th1)-like cells, causing subsequent elevations in Th1-associated inflammatory mediators, such as interleukin (IL)-2, interferon (IFN)-α, IFN-γ, monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-1 (MIP-1), and granulocyte-macrophage colony-stimulating factor (GM-CSF). CD4+ T helper cells also interact with APCs to initiate the development of and preserve granulomas.3 Thus, T cells undergo oligoclonal proliferation in areas of immune system activity. At this point in the disease process, lymphocyte levels are typically increased, and the ratio of CD4/CD8 cells becomes elevated in the lungs and other affected organs.
Acquired Immunity
In sarcoidosis, uptake of antigens is the most likely trigger for activation of macrophages, which are then able to produce IL-12, IL-15, IL-18, and tumor necrosis factor (TNF). Macrophage-derived cytokines contribute to the external signaling milieu that selectively pressures toward Th1 differentiation of CD4+ cells.12,13 Ultimately, a feedback loop from downstream-produced cytokine cascade induces macrophages to differentiate into epithelioid cells, which gain secretory capability, lose phagocytic capacity, and fuse to form multinucleated giant cells.3 These epithelioid cells form the cellular basis of granulomas. Also contributing to granuloma formation are Th2 cells. These cells synthesize fibronectin and CC motif ligand 18 (CCL18). Release of these mediators results in a positive feedback loop of CCL18-activated and macrophage-mediated collagen formation. Although granulomas spontaneously resolve without causing damage in most cases, this cycle leads to fibrosis in up to 25% of patients with sarcoidosis.3 As patients’ fibrosis becomes more extensive, their prognosis worsens. Although the mechanisms responsible for granuloma fibrosis have not been fully characterized, patterns of cytokines change, and Th2 cells may pressure toward an increased ratio of CD8 to CD4 cells. Less than 5% of patients die from sarcoidosis, but fibrosis leading to respiratory failure is a contributing factor in many sarcoidosis deaths.
In summary, the essential immunologic events in granuloma formation can be summarized as follows13,14: (1) antigen exposure, (2) antigen processing and presentation by macrophages, resulting in T cell immunity against the antigen, (3) T-effector cell production, (4) macrophage activation, and (5) granuloma formation.
Etiology
Both inorganic and organic environmental factors with antigenic capabilities have been implicated in the pathogenesis of sarcoidosis. Early studies on the causes of sarcoidosis suggested a link between sarcoidosis and agents associated with a rural lifestyle, such as the lumber industry and burning wood.15,16 These data have been extended in the ACCESS study, which found that agricultural debris and wood burning are associated particularly with pulmonary sarcoidosis but not with systemic sarcoidosis.17 In a different analysis of the ACCESS trial, radiation, insecticides, mildew, and mold were environmental factors associated with systemic sarcoidosis phenotype.18 These findings may indicate that each of these unique sarcoidosis subtypes has its own causes.
Numerous methods have been used to look for an infectious agent as a cause of sarcoidosis. When in situ hybridization was used, Mycobacterium tuberculosis catalase-peroxidase protein (mKatG) was found in nearly 40% of tissue samples from patients with sarcoidosis. Recombinant mKatG protein was then used to measure mKatG antibodies in patients with sarcoidosis, which were present in 50% of patients studied.19 Others have found evidence of an immunologic reaction to additional mycobacterial antigens.20,21 Propionibacterium acnes has been found more frequently in granulomas from sarcoidosis patients but can also be found in individuals without sarcoidosis, so its primary role in pathogenesis remains unclear.22–24 These studies suggest that sarcoidosis may represent overexposure of the patient to a commonly encountered microorganism associated with dysregulated resultant immune responses. Because several types of bacteria have been associated with sarcoidosis, some clinicians have attempted to use antibiotics to manage the disease. Although skin sarcoidosis has, on occasion, responded to antibiotics,25 their usefulness in other forms of sarcoidosis appears minimal.22
Genetics
Compelling studies of familial clustering and incidence of sarcoidosis among different racial groups indicate that sarcoidosis susceptibility is influenced by the interplay of genetic factors. In the ACCESS study, first-degree relatives were reported to have a fivefold increase in risk of developing the disease.26 Associations have been found between risk of sarcoidosis and class I and II human leukocyte antigen (HLA) gene products, which have essential roles in antigen presentation. It is considered likely that a susceptibility locus for sarcoidosis exists within the HLA gene region, as is the case with other autoimmune diseases and cancers such as Hodgkin’s lymphoma. An intriguing analysis of data from the ACCESS study identified associations between genetic factors (HLA alleles), environmental factors, and sarcoidosis phenotypes. In considering together several of the factors postulated to cause sarcoidosis, a compelling argument can be made for a genetic factor that predisposes individuals to the disease and a subsequent environmental exposure that triggers onset of sarcoidosis. Specifically, Rossman and colleagues found that HLA-DRB1*1101 and insecticide exposure at work are significantly associated with cardiac sarcoidosis and hypercalcemia.27 A similar relationship between HLA-DRB1*1101, mold and musty odors, and pulmonary sarcoidosis was described.27
HLA polymorphisms have also been linked to Löfgren’s syndrome, an acute form of sarcoidosis characterized by bilateral hilar lymphadenopathy (BHL), erythema nodosum (EN), fever, and periarticular ankle inflammation or arthritis of the ankle. In patients with Löfgren’s syndrome, HLA-DRB1*03 is four times more common than in healthy individuals28 and has been associated with EN and ankle arthritis, which are favorable prognostic factors. A more recent study found that DRB1*03-positive and DRB1*03-negative patients have different disease courses. For example, most DRB1*03-positive patients experienced resolution of Löfgren’s syndrome within 2 years after diagnosis. By contrast, nearly half of patients without this allele had resolving disease.29 The mechanism underlying this difference in disease course remains unknown. A relationship between HLA-DRB1*03 and interferon-γ-3,3 homozygosity has been suggested in sarcoidosis. Wysoczanska and colleagues reported that when combined, these two genetic factors increase the risk of Löfgren’s syndrome, and this may be indicative of a complex gene-gene interaction underlying this sarcoidosis phenotype.30
Genetic studies of non-HLA genes have been inconclusive. Loci coding for TNF, co-stimulatory molecules on antigen-presenting cells such as CD80 and CD86, chemokine receptors CCR2 and CCR5, and many others have been suggested as possible susceptibility factors, but their roles have not been fully characterized.22,31 Of these, chemokine receptor genes have been associated with particular sarcoidosis phenotypes. For example, the C-C chemokine receptor 2 (CCR2) haplotype 2 has been linked to Löfgren’s syndrome.32 In this study, the association between CCR2 haplotype 2 and Löfgren’s syndrome remained significant even after adjustment for the presence of DRB1*03.32
Ongoing genome-wide association studies (GWASs) are seeking to identify additional genes that may be linked to sarcoidosis onset and susceptibility. To date, these studies have identified several novel gene candidates.33 In family clusters with sarcoidosis, GWASs have identified areas of interest in a German population.34 A similar analysis of African-American familial sarcoidosis did not have identical findings but did find the same linkage at chromosomes 1p and 9q.35
Diagnosing Sarcoidosis
One can never be sure of the diagnosis of sarcoidosis. The American Thoracic Society (ATS), the European Respiratory Society (ERS), and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) have developed diagnostic criteria. The patient with appropriate clinical presentation and multiple organ involvement who has granulomas identified in one or more organs, and who has no other cause for the granulomatous reaction, is considered to have sarcoidosis.1 Figure 117-1 presents some features consistent with the disease.36 Diagnosis relies heavily on the finding of granulomas in the biopsy. However, many other conditions can lead to a granulomatous reaction, as is summarized in Table 117-1. Although sarcoidosis granulomas tend to be noncaseating and non-necrotizing, a significant number of sarcoidosis patients exhibit evidence of some necrosis in part of their granulomatous response. Certain features such as erythema nodosum, BHL, gallium scan showing increased activity in the parotids and lacrimal glands, an elevated angiotensin-converting enzyme (ACE) level, and bronchoalveolar lavage with increased lymphocytes with a CD4/CD8 ratio greater than 3.5 are strongly suggestive of sarcoidosis, even in a patient in whom a biopsy has not been performed.
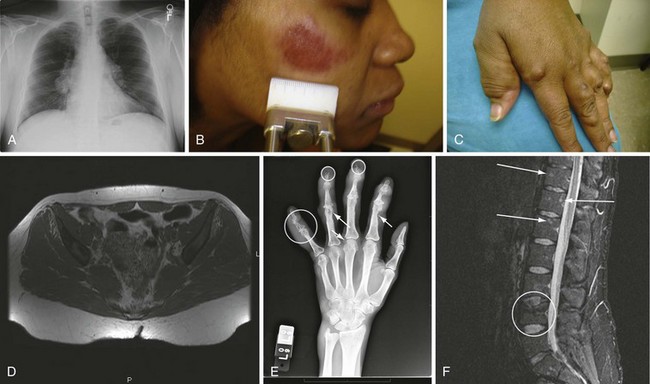
Figure 117-1 Various manifestations of sarcoidosis. A, Bilateral hilar adenopathy and right paratracheal lymph node enlargement demonstrated on a posterior anterior chest roentgenogram, Scadding stage 1.38 B, Facial lesion consistent with lupus pernio.60 C, Hand changes consistent with sarcoidosis in the fingers. D, Noncontrast magnetic resonance image of the pelvis demonstrating bone marrow replacement by granulomatous tissue. E, Cystic changes (arrows) within the bones of the fingers of a patient with sarcoidosis. F, Gadolinium enhancement of lesions of the spine seen on magnetic resonance image of a sarcoidosis patient.
(B, Reproduced with permission from the patient.)
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


