59 Prostanoid Biology and Its Therapeutic Targeting
History
Botanicals containing salicylates have been used since antiquity to treat pain, inflammation, and fever. The Egyptian Ebers papyrus recommended use of a decoction of dried myrtle leaves to be applied to the abdomen and back for relief of rheumatic pains about 3500 years ago. A thousand years later, Hippocrates recommended poplar tree juices for eye disease treatment and willow bark to alleviate fever and the pain of childbirth. Throughout Roman times, the use of botanical treatments including willow bark for pain and inflammation was widespread. The medicinal use of salicylate-containing plants occurred in China and other parts of Asia. In addition, the curative effects of other botanicals were known to the indigenous populations of North America. Colchicine-containing extracts of the autumn crocus plant were used for treatment of acute gout as early as the sixth century ad.1
The first modern report of the therapeutic application of salicylate-containing plants was reported to the Royal Society of London by the Reverend Edward Stone, who provided an account of the success of the dried bark of the willow for fever.1 In this first “clinical trial,” a pound of bark was dried, pulverized, and put into the tea, beer, or water of 50 people with fever. He found that one dose (1 dram = 1.8 g) cured their fever. In 1763 Stone wrote, “I have no other motives for publishing this valuable specific, than that it may have a fair and full trial in all its variety of circumstances and situations, and that the world may reap the benefits accruing from it.”
In 1860 salicylic acid was chemically synthesized, which led to its widespread use as an external antiseptic, antipyretic, and analgesic.1 The bitter taste of salicylic acid prompted the chemist Felix Hoffman to synthesize the more palatable acetylsalicylic acid (ASA). After demonstration of its anti-inflammatory effects, Dr. Heinrich Dreser of Bayer introduced this compound into medicine in 1899 as aspirin and it remains the most widely used drug in the world.1 Salicylate was identified as the active ingredient of willow bark in 1929.
Phenylbutazone came into clinical practice in 1949 and was followed by indomethacin, fenamates, naproxen, and others. Despite the diversity of their chemical structures, these drugs shared therapeutic properties with aspirin. Furthermore, adverse events including gastric upset, GI ulceration and bleeding, hypertension, edema, and renal damage were shared by all these drugs. In 1971 it was discovered that these drugs all acted by inhibiting PG biosynthesis, thereby providing a unifying explanation of their therapeutic actions and a rationale for grouping them together as NSAIDs.1
COX was isolated in 1976 from the endoplasmic reticulum of PG-forming cells.2,3 However, several groups speculated that there must be a second COX enzyme on the basis of observed biology. In 1990 investigators demonstrated that bacterial lipopolysaccharide (LPS) increased PG synthesis in human monocytes in vitro and in mouse peritoneal macrophages in vivo, but only the LPS-induced increase was inhibited by dexamethasone and required the de novo synthesis of “new” COX protein.4 This observation was the foundation of the concept for “constitutive” and “inducible” forms of COX. Soon thereafter a number of investigators working in different systems reported the discovery of an inducible second form of COX.3 Investigators went on to clone the gene and deduced its structure, and they found the gene product was homologous to COX, but to no other known protein. The observation that glucocorticoids inhibited the expression of COX-2 following a proinflammatory stimulus represented a link between the anti-inflammatory actions of NSAIDs and corticosteroids.
Because of the prediction that inhibiting COX-2 would block PG biosynthesis participating in the inflammatory response but was not required for homeostasis, there was a tremendous push to develop drugs that specifically inhibit COX-2 without effect on COX-1 in the belief that these medications would provide clinical efficacy without adverse effects.2,5 Identification of new drugs that differentially inhibited COX-2 over COX-1 was accomplished quickly as existing NSAIDs were tested on the two COX isoforms and crystal structures revealed differences in the protein structures on which new drug development could be based.5,6
One hundred years after aspirin was introduced and 10 years after the discovery of COX-2, selective COX-2 inhibitors, celecoxib (Celebrex) and rofecoxib (Vioxx), were developed. In clinical trials, the safety and efficacy profiles of these and related drugs showed promise and the U.S. Food and Drug Administration (FDA) subsequently approved these COX-2-selective NSAIDs for treatment of arthritis and pain. After the introduction into clinical practice, however, it became clear that the most highly COX-2-selective NSAIDs, particularly rofecoxib, were more likely than traditional NSAIDs to be associated with adverse cardiovascular events.7 This finding led to the voluntary withdrawal of rofecoxib and several other COX-2-selective NSAIDs from the market. Debate surrounding the relative risks of different NSAIDs to specific organ systems continues to the present.
Cyclooxygenase Biology and Bioactive Lipids
Therapeutic and adverse effects of NSAIDs are best understood in the context of COX biology. The COX enzymes are the first committed step in the synthesis of PG from arachidonic acid (AA) (Figure 59-1). AA is an omega-6 polyunsaturated fatty acid (PUFA) commonly found at the sn-2 position of cell membrane glycerophospholipids and cleaved from cell membranes by one of several different phospholipase A2 enzymes.8 Once generated, AA may enter several different pathways to generate bioactive lipids. It can be metabolized by COX to PGG2 and then PGH2 via its COX and peroxidase enzyme activities; by lipoxygenases (LOX) to hydroxyeicosatetraenoic acids (HETEs), hydroperoxyeicosatetraenoic acids (HPETEs), or leukotrienes; or by the cytochrome p450 family of enzymes to HETEs, HPETEs, or epoxyeicosatrienoic acids (EETs).9,10 These bioactive lipids have diverse biologic activities in normal physiology and in pathologic conditions including inflammation, pain, cardiovascular disease, and cancer.
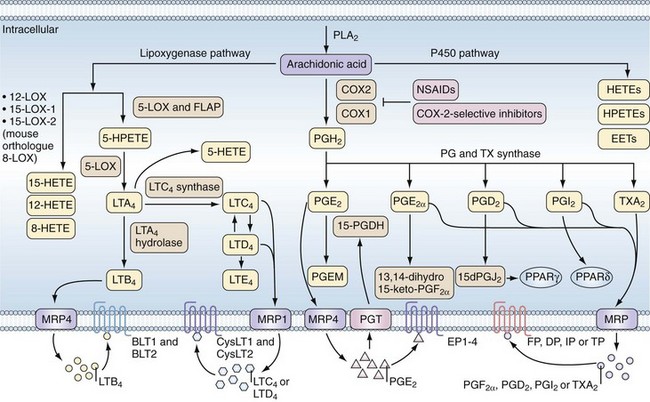
In the COX pathway, the unstable intermediate PGH2 spontaneously rearranges or is enzymatically converted by specific synthases to biologically active PG, of which there are many isoforms.3 Although phospholipase A2 activity is required to initiate PG synthesis, the overall regulation of the type and amount of PG produced in a given cell or tissue is determined by the expression levels of COX-1, COX-2, and terminal synthase enzymes.
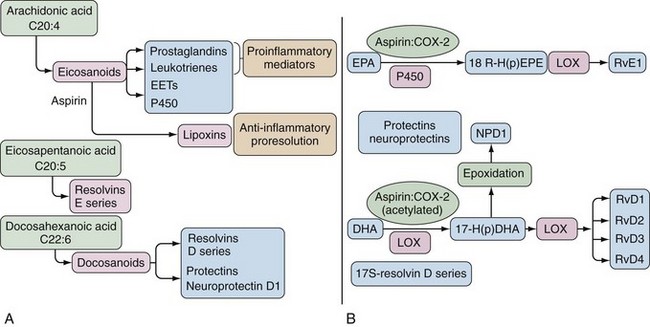
Bioactive lipids synthesized via the COX and LOX pathways are important mediators of inflammation, but alternate substrates and pathways can generate anti-inflammatory lipids and lipids important in the resolution of inflammation (Figure 59-2).11,12 Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), which are omega-3 PUFAs, can also serve as substrates for COX and LOX and their metabolic pathways lead to generation of resolvins, protectins, and maresins, bioactive lipids associated with resolution of inflammation.12,13 It is hypothesized that generation of anti-inflammatory lipids and lipids that accelerate resolution of inflammation may explain why diets high in omega-3 fatty acids are associated with reduced inflammatory and cardiovascular diseases.11 In addition, ASA possesses the capacity to alter COX-2 activity such that an “ASA-triggered” alternative catalytic cascade also results in synthesis of anti-inflammatory products including lipoxins, protectins, and resolvins (see Figure 59-2).11
Prostaglandin Production and Action
PGs act as autocrine and paracrine mediators with effects limited to the immediate vicinity of their synthesis. That PG actions are limited to their site of synthesis reflects their ephemeral nature and predisposition to metabolic inactivation to regulate promptly their potent biologic activities. Thus the enzymes required for production of a specific prostanoid must be co-localized and adjacent to the receptor mediating the action of prostanoids. Most cell types produce only one or a few prostanoid species; however, transcellular metabolism of unstable intermediates of COX may mediate qualitative and quantitative changes in the profile of eicosanoids produced in a given tissue.14 PGs are exported from cells via the multidrug resistance–associated protein family of efflux transporters, and the PG transporter mediates influx of PG and metabolism to stable products via hydroxyprostaglandin dehydrogenase (see Figure 59-1).10
PGE2, the most abundant PG at sites of inflammation, can be produced by many different cell types via at least three different PGE synthases.15 Expression of the inducible form of PGE synthase, microsomal PGE synthase-1 (mPGES-1), is similar to expression of COX-2. mPGES-1 acts in concert with COX-2 to produce high levels of PGE2 during inflammation. mPGES-1 expression is blocked by NSAIDs, and experiments demonstrate that PGE2 itself, along with proinflammatory stimuli, is required for mPGES-1 upregulation.16 This positive regulatory loop contributes to the high levels of PGE2 at sites of inflammation and likely to the efficacy of NSAIDs. Other terminal PG synthases are not known to be highly regulated, and levels of other important bioactive PGs are stably produced or their levels are increased when phospholipase activity and COX-2 levels are increased.
The actions of PG are mediated by cell surface G protein–coupled receptors (GPCRs). There are at least nine known PG receptors with additional splice variants (see Figure 59-1).17 The PG receptors belong to three clusters within a distinct subfamily of the GPCR superfamily with the lone exception of one of the PGD2 receptors (DP2), which belongs to the chemokine receptor subfamily. The relaxant receptors for prostacyclin (IP), PGD2 (DP1), and PGE2 (EP2 and EP4) signal through Gs-mediated increases in intracellular cyclic adenosine monophosphate (cAMP). The contractile receptors for thromboxane A2 (TP), PGF2α (FP), and PGE2 (EP1) signal through a Gq-mediated increase in intracellular calcium. An inhibitory receptor for PGE2 (EP3) couples to Gi and decreases cAMP formation. Note that PGE2 has at least four different receptors with a broad range of potential actions. EP4 in particular appears to mediate many of the proinflammatory activities of PGE2.18 Given the great diversity of PG receptors expressed by different cell types, PG signaling pathways constitute an enormously complex network controlling many biologic actions. Much work remains to understand fully all of the cellular signaling mechanisms by which PGs and their receptors elicit their respective biologic actions. This is particularly true as antagonists for many of these receptors show promise as novel targets for drug development.18
Biochemistry and Structural Biology
COX-1 and COX-2 are bifunctional enzymes that mediate a COX reaction whereby arachidonate plus two molecules of O2 are converted to the cyclic endoperoxide PGG2, followed by a hydroperoxidase reaction in which PGG2 undergoes a two-electron reduction to PGH2.8 COX enzymes are integral membrane proteins that sit within the inner leaflet of the lipid bilayer of intracellular phospholipid membranes of the nuclear envelope and the endoplasmic reticulum. The crystal structures of COX-1 and COX-2 have been solved, and they have essentially identical domain structures.6 The COX enzymes are homodimers with each monomer consisting of three structural domains. The N-terminal, epidermal growth factor–like domain is involved in dimerization via hydrophobic interactions. The membrane-binding domain is composed of four amphipathic α-helices lodged into half of the lipid bilayer to form a hydrophobic channel in the center of the large catalytic domain that contains the COX and peroxidase active sites and that constitutes about 80% of the protein. The catalytic domain is globular with two distinct intertwining lobes. The interface of these lobes creates a shallow cleft on the upper surface of the enzyme where the peroxidase active site is located and where heme is bound.
COX and hydroperoxidase reactions occur at distinct but structurally and functionally interconnected sites. The COX reaction is peroxide dependent and requires that the heme group at the peroxidase site undergo a two-electron oxidation. A tyrosine residue (tyrosine 385) located at the COX active site is involved as a reaction intermediate. The physiologic heme oxidant in vivo is not known, but it has been shown that the COX activity of COX-2 can be activated at 10-fold lower concentrations of hydroperoxide than that of COX-1.8
NSAIDs function by blocking access of AA to the COX active site within a long, narrow, dead-end, hydrophobic channel whose entrance is framed by the four amphipathic helices of the membrane-binding domain. The channel extends into the globular catalytic domain and is about 8 Å wide. Significant narrowing of the channel occurs where arginine 120 protrudes into the channel. Arginine 120 is essential for binding both AA substrate and most carboxylate-containing NSAIDs in COX-1. By virtue of other differences in the hydrophobic channel, this residue is unessential for binding AA in COX-2, whereas it remains critical for binding of most carboxylate-containing NSAIDs.19,20 Serine 530 is required for inhibition of COX by the phenylacetic acid NSAID diclofenac. Serine 530 is also the residue transacetylated by ASA and, along with valine 349, seems to govern the stereochemistry of the pocket such that after exposure to aspirin, AA is sterically blocked from a functional interaction with the catalytic domain in COX-1 and catalytic activity is completely inhibited.
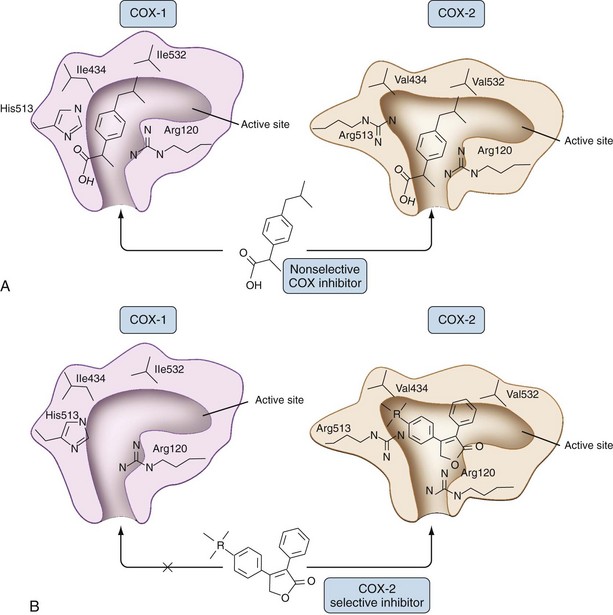
A crucial structural difference between COX-1 and COX-2 is the substitution of the small amino acid valine in COX-2 for the isoleucine with a bulky side chain in COX-1 at position 523 that opens a side pocket in the hydrophobic channel in COX-2.6 Overall, COX-2 has a wider and somewhat more flexible interior channel, a structural feature that has been exploited with respect to the development of COX-2-selective NSAIDs as shown in Figure 59-3.21
Both enzymes are homodimers, but the monomers often behave asymmetrically as conformational heterodimers during catalysis and inhibition.22 That is, when a fatty acid binds to one monomer, the other monomer becomes catalytically active and only one monomer is catalytically active at any one time. The specific fatty acid bound to the noncatalytic monomer can regulate catalytic activity.23 Different NSAIDs interact differently with respect to allosteric inhibition of COX enzymes as one facet of their pharmacology.22 ASA acetylation occurs in only one of the two COX monomers, which completely inhibits the activity of COX-1. However, COX-2 retains the ability to form a reduced amount of PGH2 and alternate aspirin-triggered lipoxins from AA. The anti-inflammatory resolvins may be synthesized by ASA-acetylated COX-2 from omega-3 PUFA.22
Molecular Biology
In addition to the differences in their structures relevant for the pharmacology of COX-1 and COX-2 inhibition, there are physiologically relevant differences with respect to expression and regulation.2,3 Generally, COX-1 is constitutively expressed in most cells and its expression is minimally altered by inflammatory stimuli. The promoter region of COX-1 has the characteristics of a gene that is continuously transcribed and stably expressed. COX-1 activity is regulated by substrate (AA) availability. When there is an increase in substrate mobilization via phospholipase A2 activation, there is a concordant increase in PG synthesis mediated by COX-1. COX-1 is the only isoform expressed in mature platelets and is the most highly expressed COX isoform in normal gastroduodenal mucosa.24 Because COX-1 is inhibited by nonselective NSAIDs, these physiologic properties may explain some of the common adverse effects of these drugs such as bleeding and GI ulceration.
In contrast, the COX-2 gene has the structure of a highly regulated product with binding sites for transcription factors such as nuclear factor κB (NFκB), cAMP-responsive element, and activating protein-1 (AP-1), which rapidly increase transcription in response to inflammatory signals.2 COX-2 expression is highly induced by proinflammatory cytokines such as tumor necrosis factor and interleukin-1β (IL-1β), microbial products, and mitogens2,8 and is inhibited by glucocorticoids.2 COX-2 mRNA stability is a key regulator of COX-2 levels. The potential for instability of the COX-2 message is due to the presence of multiple AUUUA instability sequences in the 3′ region that mediate a rapid degradation of mRNA, which ultimately suppresses COX-2 protein synthesis and PG production. Conversely, some stimuli including IL-1β may interfere with mRNA degradation and increase COX-2 levels and PG production.25 COX-1 and COX-2 can be post-translationally modified. COX-1 is glycosylated at three asparagines involved in proper protein folding of the enzyme, whereas COX-2 can be glycosylated at four asparagines.25,26
The generalization that COX-1 expression is constitutive and COX-2 expression is inducible has its limitations, given that COX-2 is expressed constitutively in several organ systems and regulated by physiologic, as well as pathologic, stimuli. COX-2 is basally expressed in the brain, kidney, pancreas, and blood vessels and therefore plays an important role in normal reproductive, renal, cardiovascular, and skeletal physiology.2,21,27
Mechanism of Action
Cyclooxygenase Inhibition
All of the NSAIDs are synthetic inhibitors of the COX active site, but subtle mechanistic differences in the manner in which individual NSAIDs interact and bind with the active site are responsible for some of the differences in their pharmacologic characteristics.28 ASA is the only covalent, irreversible modifier of COX-1 and COX-2, whereas all of the other NSAIDs are competitive inhibitors, competing with AA for binding in the active site. The competitive inhibitors are subdivided further on the basis of whether they bind to the COX active site in a time-dependent or time-independent manner.
Crystallographic studies have shown how ASA effectively acetylates serine 530 of COX-1. Similar to other NSAIDs, ASA diffuses into the COX-1 active site at the mouth of the channel and travels to the constriction created by arginine 120, where it is in the best orientation to transacetylate serine 530, leading to the complete and irreversible inhibition of COX-1.29 In COX-2, the channel of the active site is larger than COX-1, the orientation of ASA for serine 530 attack is not as good, and transacetylation efficiency for COX-2 is 10-fold to 100-fold less than for COX-1. ASA can also “trigger” COX-2 to alter its catalytic activity to produce 15 R-HETE and lipoxins from AA and to generate anti-inflammatory lipids from omega-3-PUFA.11
The time it takes for an NSAID to inhibit the COX active site relative to how long it takes for it to leave the COX channel is a crucial factor in the inhibition of COX.30 Drugs such as ibuprofen exhibit such rapid rates that they essentially inhibit COX instantly but can be removed from the COX active site just as quickly when drug levels decrease. Both COX monomers must be inhibited by ibuprofen to block catalytic activity.22 Conversely, indomethacin and diclofenac are time-dependent allosteric inhibitors that require seconds to minutes to bind to the COX active site and need only block one of the COX monomers to completely inhibit catalytic activity.22 These NSAIDs also need hours to exit the COX active site. Initially, most traditional time-dependent NSAIDs form a loose complex with the COX active site before a stronger interaction is established. This complex is limited by the time it takes the drug to become properly oriented within the COX channel at arginine 120, the constriction site in the COX channel. This may involve a change in conformation to the “open state” to allow the drug to access the upper part of the COX catalytic site.
Drugs such as flurbiprofen and indomethacin form a salt bridge between the carboxylate moiety of the NSAID and the guanidinium moiety of arginine 120. Hydrophobic interactions between the aromatic rings and the hydrophobic amino acids in the channel aid binding. Such interactions at the constriction point of the channel completely block the entry of substrate to the active site.31 Diclofenac interacts with serine 530, not arginine 120, but also blocks entry of substrate.32
COX-2 Selectivity
NSAIDs such as meloxicam, nimesulide, and etodolac show some selectivity for inhibiting COX-2 over COX-1. After the discovery of COX-2, efforts to further enhance COX-2 selectivity led to the development of celecoxib, rofecoxib, valdecoxib, etoricoxib, and lumiracoxib. The prototypical COX-2-selective NSAIDs, celecoxib and rofecoxib, are diaryl compounds containing a sulfonamide (celecoxib) and methylsulfone (rofecoxib) rather than a carboxyl group. Both drugs are weak time-independent inhibitors of COX-1 but strong time-dependent inhibitors of COX-2 that require their entry into and stabilized binding in the catalytic pocket. Because these drugs lack a carboxyl group, arginine 120 is not involved, but multiple sites of hydrogen and hydrophobic binding stabilize drugs at the catalytic site. The sulfur-containing phenyl ring of COX-2-selective NSAIDs plays a pivotal role in binding stability by occupying the hydrophobic side pocket characteristic of the COX-2 catalytic site. If this side pocket is removed by mutagenesis, all isozyme selectivity is lost.6
COX isozyme selectivity is defined most commonly using the concentration of drug required to inhibit PG production by 50% in a particular assay system (inhibitory concentration, or IC, 50). Ratios using values obtained for COX-1 IC50s compared with COX-2 IC50s can be calculated and used as a standard measure for comparing the degrees of selectivity of a particular NSAID for one or the other COX isoform.33 PG assay systems can vary widely, however, making it difficult to compare directly results from studies using different assay systems. To circumvent such problems, most clinicians have accepted the use of the in vitro whole-blood assay to compare NSAID selectivities. In this system, COX-1 inhibition is assessed as a function of the reduction of thromboxane made by platelets after clot formation. Inhibition of COX-2 is based on the inhibition of PGE2 production in a heparinized blood sample after LPS stimulation. A COX-2-selective NSAID lacks inhibitory effect on platelet COX-1 at concentrations at or above those that maximally inhibit COX-2.5,34
Cyclooxygenase-Independent Mechanisms of Action
At high, nonphysiologic concentrations, some NSAIDs seem to elicit effects on cellular pathways in vitro that do not involve the inhibition of COX. Because of the high doses of drug required and the use of in vitro systems, the relevance of these effects to in vivo activity is uncertain. Some NSAIDs inhibit phosphodiesterases associated with the metabolism of cAMP leading to increased intracellular cAMP levels and the subsequent general inhibition of peripheral blood lymphocyte responses to mitogen stimulation, monocyte and neutrophil migration, and neutrophil aggregation.35 NSAIDs scavenge free radicals, inhibit superoxide production by polymorphonuclear neutrophils, reduce mononuclear cell phospholipase C activity, and inhibit inducible nitric oxide synthase activity. Sodium salicylate and ASA inhibit NFκB activation, as do certain inactive enantiomers of flurbiprofen. Some reports indicate that other cell signaling molecules such as mitogen-activated protein kinases and the transcription factor AP-1 may also be modulated by NSAIDs. Some NSAIDs bind to and activate members of the peroxisome proliferator-activated receptor (PPAR) family and other intracellular receptors. PPAR activation is thought to mediate anti-inflammatory activities. Selective COX-2 inhibitors may have unique structural features that promote COX-independent activities such as cell-cycle regulation, apoptosis, and antiangiogenesis.36
Mechanism of Acetaminophen and Other Analgesic Antipyretic Drugs
Acetaminophen (paracetamol) and dipyrone relieve pain and fever, but they are not anti-inflammatory. The precise mechanisms by which these drugs elicit their effects remain unclear. In the 1970s, it was proposed that acetaminophen worked by means of a “central” action by inhibiting COX activity primarily in the brain and not in peripheral tissues because they were not acidic and could cross the blood-brain barrier.37 Acetaminophen does inhibit COX-1 and COX-2, but variably so and dependent on cell and tissue type. Acetaminophen does not appear to inhibit by interaction with the COX active site; rather, it serves as a reducing co-substrate for the peroxidase site. The peroxide tone of cells and tissues in vivo may be responsible for inhibitor specificity, with platelets and activated macrophages being resistant to the action of acetaminophen and vascular endothelial cells being sensitive to its inhibitory effects on COX. Additionally, the inhibitory potency of acetaminophen is determined by the concentration of the COX enzyme.37 This may be an additional factor for the lack of clinical anti-inflammatory effects because inflammation is associated with a markedly increased expression of COX-2 enzyme. With the discovery of a COX-1 splice variant and studies showing that it is both highly expressed in brain and more sensitive to inhibition by acetaminophen, some authors proposed that the analgesic and antipyretic actions of acetaminophen could be explained by its ability to inhibit the COX-1 splice variants (called COX-3 by some despite the fact that this variant does not arise from a unique gene).38 However, more recent studies have rejected this mechanism as explanatory for acetaminophen effects.37,39
Salicylate has analgesic, antipyretic, and anti-inflammatory activity but, similar to acetaminophen and in contrast to ASA, is a poor COX inhibitor. Salicylate has also been shown to inhibit COX activity if substrate levels are low, and it is also dependent on the oxidative state of the enzyme, suggesting that this drug may inhibit COX by redox-related mechanisms.40
Pharmacology and Dosing
Classification
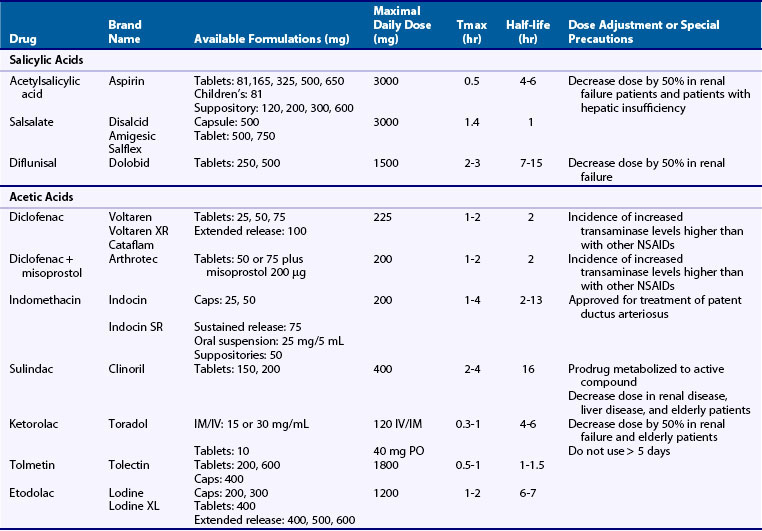
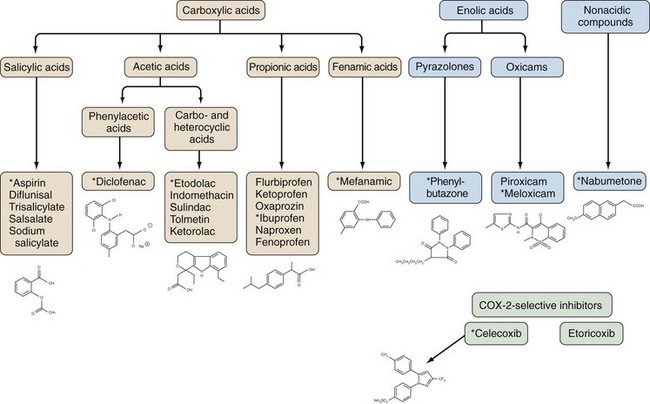
NSAIDs are generally grouped according to their chemical structures, plasma half-life, and COX-1 versus COX-2-selectivity (Table 59-1 and Figure 59-4). Table 59-1 presents a representative compilation of common NSAIDs, formulations, dosages, half-lives, and precautions. Structurally, most NSAIDs are organic acids with low pK values that lend themselves to their accumulation at sites of inflammation, areas that often exhibit lower pHs than uninvolved sites. Most often, there is a direct relationship between low pK and short half-life, but there are exceptions such as nabumetone, which is nonacidic. Classifying NSAIDs on the basis of plasma half-life can be problematic given the fact that these drugs tend to accumulate in synovial fluid, where the concentration of drug may remain more stable than in the plasma. Short half-life NSAIDs potentially could be given less frequently than indicated by their plasma half-life. NSAIDs exhibiting longer half-lives require more time to reach steady-state plasma levels. Drugs with a half-life greater than 12 hours can be given once or twice a day. Plasma levels increase for a few days to several weeks (depending on the specific half-life) but then tend to remain constant between doses. NSAIDs with longer half-lives also enable drug concentrations to equilibrate between the plasma and the synovial fluid, though total bound and unbound drug levels are usually lower in synovial fluid because there is less albumin in synovial fluid than in plasma. However, NSAIDs with longer half-lives or extended release formulation may be associated with increased propensity to cause adverse effects.41 COX-isozyme selectivity is likely to be a critically important factor in determining relative GI and cardiovascular risk, which should also be considered in addition to other pharmacologic properties for each NSAID.33
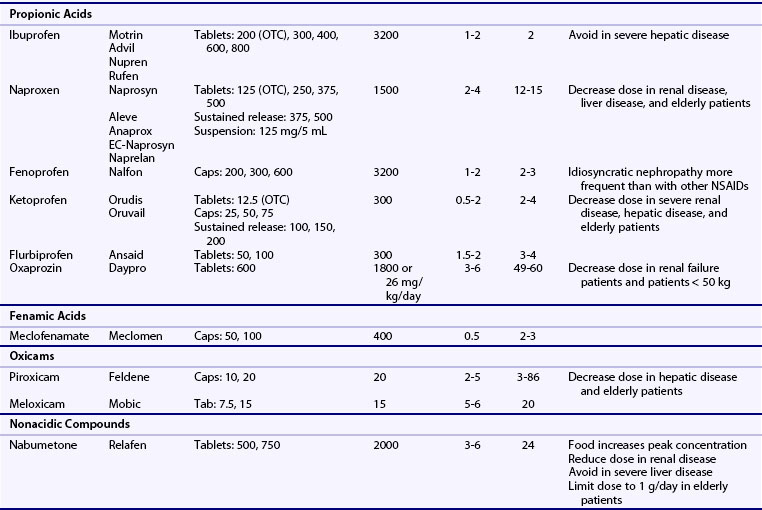

Salicylate Metabolism and Aspirin Resistance
Salicylates are acetylated (e.g., aspirin) or nonacetylated (e.g., sodium salicylate, choline salicylate, choline magnesium trisalicylate, salicylsalicylic acid).40 Although the nonacetylated salicylates are only weak inhibitors of COX in vitro, they are able to reduce inflammation in vivo. Aspirin is rapidly deacetylated to salicylate, both spontaneously and enzymatically. Differences in formulation of these agents affect the absorption properties, but not bioavailability. Buffered aspirin tablets contain antacids that increase the pH of the microenvironment, whereas enteric coating slows absorption. The bioavailability of rectal aspirin suppositories increases with retention time. Salicylates primarily bind to albumin and rapidly diffuse into most body fluids. Salicylate is metabolized principally by the liver and excreted primarily by the kidney. In the kidney, salicylate and its metabolites are freely filtered by the glomerulus, then reabsorbed and secreted by the tubules. Salicylate serum levels usually do not correlate well with dosage, however, and small increases in dosage may result in disproportionate increases in serum levels. The drug clearance rate is a function of serum concentration. The primary factors regulating serum salicylate levels are urinary pH and metabolic enzyme activity.
The term “aspirin resistance” is broadly used to describe the failure of aspirin to prevent a thrombotic event whether due to pharmacologic resistance to the antiplatelet effects of aspirin or due to the inability of aspirin to overcome thrombophilia in a given clinical setting.42 Factors such as sex, genetic polymorphisms, and clinical factors including smoking, obesity, and diabetes may alter aspirin effects on platelet function. Lack of adherence and drug interactions also may play a role in aspirin resistance.
Pharmacologic Variability
Different patients can respond to the same NSAID in a variety of ways, and the basis for this individual variability remains unclear. Several pharmacologic factors related to NSAIDs may influence this variability such as dose response, plasma half-life, enantiomeric conversion, urinary excretion, and pharmacodynamic variation.43 Other important drug factors include protein binding, the metabolic profile of the drug, and the percentage of the drug that is available as the active (S) enantiomer. Some NSAIDs exist as two enantiomers; these include the propionic acid derivatives ibuprofen, ketoprofen, and flurbiprofen, which exist as mixtures of inactive (R) and active (S) enantiomers. Naproxen is composed of the active (S) enantiomer. Conversion of the propionic acid NSAIDs from the inactive (R) enantiomer to the active (S) enantiomer occurs in vivo to various degrees, providing some basis for the variability in patient response. There is also genetic variability in the cytochrome P450 metabolic enzymes such that some individuals or ethnic groups metabolize drugs more slowly. For example, Asians are frequently slow metabolizers through the CYP2C9 pathway. Finally, the pharmacokinetics of some NSAIDs are affected by hepatic disease, renal disease, or old age.
Routes of Drug Delivery
Topical NSAID formulations were developed to reduce systemic exposure while preserving efficacy. Diclofenac, for example, is available as a solution, gel, or patch. The systemic effects are directly proportional to the surface area, and this method of delivery results in a relatively stable systemic diclofenac level compared with oral administration.44
Combination Drugs
NSAIDs have also been combined with agents having gastroprotective effects into “polypills” that are currently available on the market. This strategy may increase compliance with effective protective agents, thereby reducing adverse effects in clinical practice. Combining diclofenac with the synthetic PGE1 analogue misoprostol (Arthrotec) is shown to reduce risk of NSAID-related peptic ulcerations and mucosal injury, but utility of the combination is often limited by misoprostol-induced cramping and diarrhea.45 In population-based studies, Arthrotec was more effective than diclofenac and misoprostol co-prescription in preventing hospitalization for peptic ulcer disease or GI hemorrhage.46 The combination of enteric-coated naproxen and the proton pump inhibitor (PPI) esomeprazole (Vimovo) into a single pill has been approved by the U.S. Food and Drug Administration. This agent was shown to reduce endoscopically detected gastric ulcers.47
A different strategy is nitric oxide releasing NSAIDs (NO-NSAIDs), which are synthesized by the ester linkage of an NO-releasing moiety to conventional NSAIDs including aspirin, flurbiprofen, diclofenac, sulindac, and others.48 The NO moiety is slowly released by enzymatic activity in vivo, likely by esterases, resulting in slow accumulation of the parent NSAID. The lower rate of GI ulceration associated with these drugs is likely related to NO-associated vasodilation and the relatively lower concentration of the parent NSAID.
Therapeutic Effects
Anti-inflammatory Effects
NSAIDs are frequently used as first-line agents for the symptomatic relief of many different inflammatory conditions. In double-blind, randomized clinical trials of inflammatory arthritis, NSAIDs have been compared with placebo, aspirin, and each other. Clinical trials of NSAID efficacy in rheumatoid arthritis (and osteoarthritis) most often employ a design whereby the current NSAID is discontinued and the patient must have an increase in symptoms or flare to enter the study. Although there is some variation in primary outcome measures, most include parameters that make up the American College of Rheumatology (ACR)-20. Efficacy superior to that of placebo is easily demonstrated for NSAIDs within 1 to 2 weeks in patients with active RA who are not receiving corticosteroids or other anti-inflammatory medications.49 Comparisons of adequate doses of traditional NSAIDs or COX-2-selective NSAIDs with one another almost always show comparable efficacy. Despite improvement in pain and stiffness with NSAIDs, these agents do not usually reduce acute phase reactants, nor do they modify radiographic progression. The anti-inflammatory effects of NSAIDs have also been demonstrated in rheumatic fever, juvenile rheumatoid arthritis, ankylosing spondylitis, gout, osteoarthritis, and systemic lupus erythematosus (SLE). Although not as rigorously proven, their efficacy is also accepted in treatment of reactive arthritis, psoriatic arthritis, acute and chronic bursitis, and tendinitis.
Analgesic Effects
Virtually all NSAIDs relieve pain when used in doses substantially lower than those required to suppress inflammation. The analgesic action of NSAIDs is due to inhibition of PG production in peripheral tissues and in the central nervous system (CNS). In the periphery, PGs do not induce pain per se but rather sensitize peripheral nociceptors to the effects of mediators such as bradykinin or histamine.50 PGs released during inflammation or other trauma lower the activation threshold of tetrodotoxin-resistant sodium channels on sensory neurons. In the CNS, where NSAIDs and acetaminophen exert analgesic effects, PGs also play an important role in neuronal sensitization. COX-2 is constitutively expressed in the dorsal horn of the spinal cord, and its expression is increased during inflammation.51 Centrally generated PGE2 activates spinal neurons and also microglia that contribute to neuropathic pain.52 Both COX-1 and COX-2 play a role in nociception as demonstrated by reductions in experimental pain in mice deficient in either COX-1 or COX-2.53
Antipyretic Effects
The NSAIDs and acetaminophen effectively suppress fever in humans and experimental animals. Fever results from the production of PG, primarily PGE2, from vascular endothelial cells via COX-2 and mPGES-1.54 These PGs generate neuronal signals that activate the thermoregulatory center in the preoptic area of the anterior hypothalamus. PGE2 synthesis is stimulated by endogenous (e.g., interleukin-1) or exogenous (e.g., lipopolysaccharide) pyrogens. Mice with a targeted deletion of either the COX-2 or mPGES-1 genes fail to develop fever in response to inflammatory stimuli.55
Little evidence suggests that any NSAID has superior efficacy as an antipyretic. However, in fever associated with viral illnesses, aspirin should be avoided due to the association with hepatocellular failure (Reye’s syndrome).56
Other Therapeutic Effects
Antiplatelet Effects
Aspirin and traditional NSAIDs inhibit platelet COX-1 to variable degrees. Except for aspirin, inhibition of platelet aggregation is reversible and depends on the concentration of drug in the platelet. Aspirin acetylates platelet COX-1, which cannot be resynthesized. The antiaggregation effect of as little as 80 mg of aspirin can last for up to 4 to 6 days, until the bone marrow can synthesize new platelets.57
On the basis of accumulated data showing its benefits, the FDA has approved ASA for use in the secondary prevention of cardiovascular disease. Major trials have shown that meaningful decreases in nonfatal myocardial infarction (MI), nonfatal stroke, and death can be realized by daily administration of ASA of 75 to 325 mg. Major vascular events can be reduced by 10 to 20 events for every 1000 patients treated, at a cost of one to two major GI bleeds.58
There was no reduction in rates of MI observed with the use of ASA, 100 mg every other day, in the Nurses Health Study of primary prevention of major vascular events, whereas rates of GI bleeding were increased. However, stroke rates were significantly reduced on this regimen.59 The U.S. Preventative Health Task Force has updated its recommendations to encourage use of low-dose ASA in men aged 45 to 79 and women aged 55 to 79.58 ASA used for primary prevention of cardiovascular events appears to lower the risk for MI in men and for stroke in women.60
Cancer Chemoprevention
A large body of epidemiologic and animal studies provides evidence that a high-fat diet can be associated with a risk for cancer. AA, one of the major ingredients of animal fats, and the eicosanoids derived from AA are shown to be important contributors to cancer development.10 Large-scale epidemiologic studies have long indicated that long-term NSAID use reduces the incidence of a variety of cancers including colon, intestinal, gastric, breast, and bladder, 40% to 50%.10 Given the ability of the NSAIDs to inhibit COX and PG production, the COX pathway immediately becomes implicated as playing an important role in the pathogenic process. It is well recognized that growth factors, tumor promoters, and oncogenes stimulate PG production via the induction of COX-2 and that human tumorigenic tissues exhibit increased COX activity compared with their normal, nontumorigenic counterparts. COX-2 is overexpressed in 80% of colorectal cancer tissues. Among the PGs, PGE2 is most abundant in human neoplasms. The inducible mPGES-1 enzyme is highly expressed in tumors, and its absence suppresses intestinal tumorigenesis in animal models. Furthermore, the enzyme that metabolizes intracellular PGE2, 15-hydroxyprostaglandin dehydrogenase, is ubiquitously lacking in tumors, and mice with a genetic deletion of this enzyme have accelerated tumorigenesis.10 Many natural products including resveratrol (red wine), catechins (green tea), and curcumin (saffron) also inhibit COX, which may be an important mechanism underlying their putative cancer-preventing effects.61
A retrospective cohort study shows that aspirin and traditional NSAIDs specifically reduce cancer risk in the subgroup of patients whose colon tumors express higher levels of COX-2.62 In a meta-analysis of ASA (75 mg daily and upward without dose dependence) effects on cancer, allocation to aspirin reduced death due to cancer by more than 20%.63 On analysis of individual patient data, cancer death benefit was apparent only after 5 years’ follow-up, and benefit increased with scheduled duration of trial treatment. ASA effects appear greater on adenomatous cancer than other cancer types. Other studies demonstrated a reduction in both incidence of colorectal cancer and death from colorectal cancer, particularly for cancers of the proximal colon.64 Long-term low-dose ASA use also appears to reduce the risk of prostate cancer.65
Clinical trials also demonstrated that traditional and COX-2-selective NSAIDs could cause regression of polyps in patients with familial adenomatous polyposis (FAP).66 Celecoxib was subsequently approved by the FDA for reduction of polyps in patients with FAP. Although NSAIDs are still among the most promising chemopreventative agents for cancer, cardiovascular and GI side effects have dampened enthusiasm.10 Alternative strategies for blocking effects of PGE2 and other eicosanoids need evaluation to determine their effectiveness as cancer prevention and adjunct therapy.
Adverse Effects
The NSAIDs share a common spectrum of clinical toxicities, although the frequency of particular side effects varies with the compound (Table 59-2). Hazard of individual NSAIDs is related to the pharmacologic characteristics such as bioavailability and half-life, as well as potency for inhibition of COX-1 and COX-2.33,41,67
Table 59-2 Shared Toxicities of Nonsteroidal Anti-inflammatory Drugs
| Organ System | Toxicity |
|---|---|
| Gastrointestinal | |
| Renal | < div class='tao-gold-member'> Only gold members can continue reading. Log In or Register to continue
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
