90 Polyarteritis Nodosa and Related Disorders
The true incidence and prevalence of nonhepatitis PAN is around 1 per million per annum.
Glomerular renal disease is absent in PAN and should prompt consideration of other diagnoses.
Hematuria with renal impairment is rare but can occur in the presence of renal infarction.
The role of tobacco use in Buerger’s disease is clearly established but not understood.
Rare forms of vasculitis are described, but the evidence base for treatment is limited.
Polyarteritis Nodosa
Definition and Classification
The term periarteritis nodosa was originally introduced in 1866 and subsequently used to describe any form of systemic vasculitis.1 The term has been modified to polyarteritis nodosa (PAN), and the definition has been improved to consist of “necrotizing inflammation of medium-sized or small arteries without glomerulonephritis or vasculitis in arterioles, capillaries, or venules.”2
The American College of Rheumatology (ACR) criteria, which can be used to classify patients as having PAN, in order to distinguish them from patients with other forms of primary systemic vasculitis, do not differentiate between PAN and microscopic polyangiitis (MPA); both conditions are included under the umbrella of PAN. Three of 10 criteria (listed in Table 90-1) are required.3
Table 90-1 American College of Rheumatology Criteria for Classification of Polyarteritis Nodosa (and Microscopic Polyangiitis)
From Lightfoot RW, Michel BA, Bloch DA, et al: The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa, Arthritis Rheum 33:1088–1093, 1990.
The sensitivity of the ACR criteria for PAN (or MPA) is 82.2%, with a specificity of 86.6% when used to classify the disease.3 Definitions of vasculitis (including PAN) were published in 1994 by the Chapel Hill Consensus Conference (CHCC).2 Microscopic polyangiitis, granulomatosis with polyangiitis (GPA) (formerly Wegener’s granulomatosis), and Churg-Strauss syndrome were distinguished from PAN on the basis of the pathologic findings of small vessel involvement in the former three diseases; by contrast, small vessel involvement is absent in PAN.
There has been, and still is, much confusion in the terms used to describe patients with different forms of vasculitis. It is worth considering this for a moment because, on reviewing patients who have previously been given the label of “PAN,” the clinical features at presentation might actually suggest another form of vasculitis, typically MPA or GPA. In an attempt to rationalize the naming of different forms of vasculitis, the European Medicines Agency (EMEA) produced an algorithm to assist in the classification of patients with systemic small and medium vessel primary systemic vasculitis.4 A decision tree approach was used, effectively putting PAN at the bottom of the tree.4 In other words, patients were assigned to any other form of vasculitis, and only as a last resort, if no other diagnosis was made, the term PAN was applied. Perhaps this might seem a rather negative view of the condition, but because of the overuse of the term PAN in preference to others in the past, the true incidence and prevalence of PAN are actually low.
The EMEA algorithm is particularly useful because MPA is a more common condition than PAN but is absent from the ACR classification criteria (it is considered part of PAN), whereas in the CHCC definition PAN and MPA are treated as separate entities. There are important differences between PAN and MPA in terms of pathogenesis, organ involvement, tendency to relapse, and prognosis.5 In the CHCC definition PAN is a medium vessel disease; by comparison, MPA is predominantly a small vessel disease that includes glomerulonephritis and pulmonary capillaritis.2
Patients are assessed using the EMEA algorithm if they have a clinical diagnosis of an antineutrophil cytoplasm antibody (ANCA)-associated vasculitis or PAN. In the algorithm, PAN is regarded as a diagnosis of exclusion. The first part of the algorithm is to determine whether the patient fulfills the Lanham6 or ACR criteria7 for Churg-Strauss syndrome (CSS). If they do, they are classified as having CSS. If not, the next step is to determine whether they fulfill the ACR criteria or CHCC definition for GPA2,8 either by direct evidence with histology or with appropriate surrogate markers and a positive ANCA. If they fulfill these requirements, they are classified as having GPA. If not, they proceed down the algorithm to see whether they fulfill a definition of MPA.2 This is either by histology showing small vessel vasculitis or glomerulonephritis and no surrogate markers for GPA or with surrogate markers of glomerulonephritis and a positive ANCA. Only when CSS, GPA, and MPA are excluded is a diagnosis of PAN possible in this schema. To fulfill the definition of PAN in this algorithm, there must be histology or angiographic features consistent with the diagnosis. Any remaining patients are determined to be unclassifiable. The initial validation of the algorithm made use of paper cases assessed by a number of experts in vasculitis. In fact, every case was assigned to one of the conditions, without the need to describe any case as unclassifiable. This may not reflect clinical practice, where some patients appear to have an overlap between different forms of vasculitis or incomplete forms of a vasculitis. This is an important issue and has started to be addressed by a recent task force on classification and diagnostic tests in vasculitis.9
The French Vasculitis Study Group proposed a set of predictive items (Table 90-2) to be used as diagnostic criteria.10 The items were derived from 949 patients with known vasculitis (including 262 described as having PAN) and not from undifferentiated patients, and therefore they are actually another form of classification criteria.
Table 90-2 Predictive Variables for Classification of Polyarteritis Nodosa (PAN)
| Variable | Positive Prediction for PAN | Negative Prediction for PAN |
|---|---|---|
| Positive hepatitis B serology | + | |
| Arteriographic abnormalities | + | |
| Mononeuropathy or polyneuropathy | + | |
| Positive antineutrophil cytoplasm antibody | + | |
| Asthma | + | |
| Ear, nose, or throat signs | + | |
| Glomerulopathy | + | |
| Cryoglobulinemia | + |
Modified from Henegar C, Pagnoux C, Puéchal X, et al: A paradigm of diagnostic criteria for polyarteritis nodosa: analysis of a series of 949 patients with vasculitides, Arthritis Rheum 58(5):1528–1538, 2008.
Epidemiology
The epidemiology of PAN has changed over time. Effective hepatitis B virus (HBV) immunization programs, improved blood screening for HBV, and major changes in definition and classification of vasculitis have resulted in a substantial reduction in incidence of PAN. Before the CHCC definition in 1994, MPA was included in the incidence and prevalence estimates. The impact of this is highlighted in a study comparing the incidence of PAN in three European regions: 4.4 to 9.7 per million by the ACR criteria versus 0 to 0.9 per million with the CHCC definition.11
The incidence of PAN is 2 to 9 per million in Europe and the United States by ACR criteria.12 A higher incidence of 16 per million (by CHCC definition) is reported in Kuwait13 and 77 per million described in an Alaskan population endemic for HBV, although it was based on only 13 actual cases of HBV PAN (in a study performed before ACR criteria or CHCC definitions).14 The prevalence of PAN by ACR criteria is 31 to 33 per million in Western Europe15–17; 2 to 9 per million in Germany by CHCC definition.18 A small study from Australia assessed the incidence and prevalence of different sorts of vasculitis between 1995 and 2005 in one area, suggesting a fall in the incidence of PAN from 2.3 per million per annum to 1.1 per million per annum.19 A study from 2009 assessed the incidence and survival of patients with different forms of vasculitis and estimated the incidence of PAN to be 0.9 (between 0 and 1.7) per million per annum. This compares with an almost 10-fold difference in incidence of GPA and MPA (between 9.8 and 10.1 per million per annum).20
PAN can occur at any age, but the commonest age range at diagnosis is 40 to 60 years. There is no clear gender difference.17 In a multiethnic population from France, patients with European ancestry had a higher prevalence of PAN compared with those without.16
Etiology and Pathogenesis
PAN is often related to infection with hepatitis B.21 The incidence of HBV-related PAN follows HBV infection rates, previously accounting for 7% to 38.5% of patients diagnosed with PAN.16,22 The prevalence of HBV-related PAN has reduced in recent years as a result of vaccination for HBV and improved screening of blood products.16,23 PAN develops in 1% to 5% of patients with HBV infection,14 which confers an approximately 1000-fold increase in risk compared with the background population.12 By contrast, in Alaska, an area endemic for HBV, there is a substantial increased annual incidence of HBV PAN (77 per million), primarily due to vertical transmission.14 Documented exposure to HBV occurs from blood products, intravenous drug use, and sexual contact. Screening of blood products for HBV and mass vaccination against the virus has successfully reduced the incidence of HBV PAN, as shown in French patients with the proportion of PAN due to HBV falling from 38.5% in the 1970s to 17.4% by the period 1997-2002.24
In HBV-related PAN the postulated mechanism of vasculitis includes direct vessel injury by replicating virus or deposition of immune complexes. Deposition of immune complexes leads to activation of the complement cascade, resulting in an inflammatory response and subsequent endothelial damage. The vasculitis typically occurs within the first few months following HBV infection and may be the first presenting feature of this infection. Evidence for the pathogenic nature of HBV and immune complexes is supported by the effectiveness of a treatment strategy to eradicate HBV with antiviral therapy and removal of immune complexes by plasmapheresis without the need for long-term immunosuppression.24,25
Saadoun and colleagues26 described 31 patients who had hepatitis C virus (HCV)-associated vasculitis, which they classified as PAN according to ACR criteria and CHCC definitions. This cohort, representing approximately one-fifth of their HCV vasculitis patients, had more frequent fever, weight loss, severe hypertension, gastrointestinal tract involvement, severe acute sensory-motor multifocal mononeuropathy, kidney and liver microaneurysms, and increased C-reactive protein compared with other HCV vasculitis patients. By contrast, their response to therapy was more successful.
In the remainder of patients with PAN, the etiology is unknown. Genetic, infectious, and environmental agents are thought to be important, but there is no conclusive evidence.11 The fact that idiopathic PAN responds to immunosuppressive therapy suggests an immunologic mechanism. In idiopathic PAN the role of immune complexes is unclear. There is evidence of endothelial dysfunction, an increase in inflammatory cytokines, and an increase in expression of adhesion molecules. There is a propensity for the inflammatory lesions to occur at the sites of bifurcation in vessels, where there is most likely to be turbulent flow. Following the inciting event, there is focal and segmental necrotizing inflammation of medium- and small-sized arteries. This leads to intimal proliferation with subsequent thrombosis, resulting in ischemia or infarction of the organ or tissue supplied by these arteries.
There are case reports describing the development of PAN in patients with pre-existing hairy cell leukemia.27,28 Most cases had undergone splenectomy before the development of PAN. Potential mechanisms for the association between hairy cell leukemia and PAN include cross-reactivity of antibodies between the tumor cells and the endothelium, direct damage of the endothelium by tumor cells, and local production of proinflammatory cytokines triggering vessel wall damage.28
Pathologic Features
Due to the protean manifestations of the disease, a number of different sites can be biopsied. Sampling should be directed by the pattern of clinical involvement suggested by the history or physical examination. Muscle, peripheral nerves, kidney, testis and rectum, when involved, provide the best yield. Skin involvement and a positive skin biopsy do not always indicate evidence of systemic involvement29–31 (see section on cutaneous PAN).
A biopsy of a medium-sized artery will show “focal and segmental” transmural necrotizing inflammation30,31 (Figure 90-1). If there are any clinical or laboratory features indicating involvement of small vessels, further assessment should be undertaken because it suggests an alternative form of vasculitis such as MPA.32
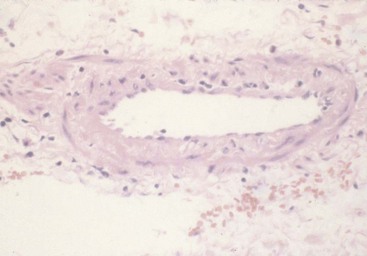
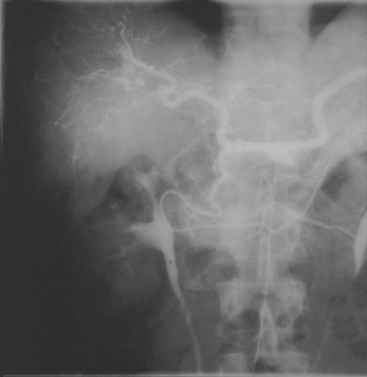
Inflammatory change at the bifurcation of vessels is reported to be common. The coexistence of different stages of inflammation and scarring, as well as normal vessel wall, is typical in PAN. Areas of acute inflammation will usually have a pleomorphic cellular infiltrate of lymphocytes, neutrophils, macrophages, and eosinophils. Aneurysms can occur at the site of active lesions, and this morphologic appearance is what led to the term “nodosa” (Figure 90-2). Proliferative scarring in other areas may lead to vessel narrowing.30–33
Clinical Features
PAN can present at any age, but it typically occurs in the age range of 40 to 60. There is no significant gender difference. The diagnosis can be difficult to make because individual features can occur in a variety of other diseases. Table 90-3 summarizes the clinical findings in PAN. The presence of nonspecific constitutional symptoms such as fever, weight loss, joint and muscle pains (found in 65% to 80% of cases of PAN), and ischemic symptoms in one or more organ system should raise the possibility of a systemic vasculitis.17,24 Organ-specific manifestations may be present at the onset of disease, or there may be a more low-grade course, resulting in the development of specific organ involvement months to years later. Involvement of organs may be in isolation or as multisystem disease. In a Swedish series, the most common manifestations occurred at disease onset: nervous system (55%), skin (44%), abdominal (33%), and renal (11%).17 A review of 348 patients diagnosed as having PAN since the 1960s34 suggested that their mean age of diagnosis was 51 and the most frequent symptoms were generalized features in more than 90% of cases, neurologic features in 79%, skin involvement in 50%, abdominal involvement in 36%, hypertension in 35%, renal artery microaneurysms in 66%, and histologically proven PAN in 70%. The subset of 123 patients with hepatitis B–associated PAN was more likely to suffer peripheral neuropathy, abdominal pain, cardiomyopathy, orchitis, and hypertension compared with 225 non-HBV PAN patients. The relapse rate was 22% over a follow-up of 6 years (28% for non–HBV-related PAN compared with 11% for HBV PAN). The mortality rate was 25% overall (20% for non-HBV PAN compared with 34% in HBV-PAN). Five-year relapse-free survival was higher in patients with HBV-related PAN (67% compared with 59%). The conclusion was that non-HBV PAN in particular had a high mortality, especially involving the elderly, and was worse in those patients with skin manifestations.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


