108 Pediatric Systemic Lupus Erythematosus, Dermatomyositis, Scleroderma, and Vasculitis
Systemic Lupus Erythematosus
Definition and Classification
Pediatric SLE (pSLE) is a chronic multisystem autoimmune disease with remitting, relapsing course and onset of symptoms before age 18 years, accounting for approximately 20% of all SLE.1 This clinically heterogeneous disease is characterized by a distinct spectrum of autoantibodies including antinuclear antibody (ANA), double-stranded deoxyribonucleic acid (dsDNA), and antibodies against extractable nuclear antigens (ENAs). In genetically susceptible hosts B cell–mediated autoimmune processes lead to a variable combination and severity of clinical symptoms including antibody-mediated vasculitis, direct antibody binding to target cells, and thrombotic organ dysfunction.
The American College of Rheumatology (ACR) classification criteria for adults with SLE are commonly applied to children with pSLE.2 The classification of neuropsychiatric SLE (NPSLE) in children and adolescents remains a challenge.3,4 The 1990 ACR NPSLE nomenclature and case definitions appear to have limited applicability for children in certain domains.5–7
Epidemiology
Pediatric SLE affects children and adolescents around the world.8–10 On average, 60% of patients develop pSLE after age 10, 35% between 5 and 10 years, and only 5% before age 5. In studies from Asia, the mean ages at diagnosis were reported to be 8.6 to 13.5 years.8,9 The incidence and prevalence of pSLE varies between populations. Similar to adults, SLE more commonly affects non-Caucasian populations.8,9,11 Overall, incidence rates of pSLE have been reported to be 0.28 to 0.48 per 100,000 children with prevalence rates of 6.3 to 24 per 100,000 depending on the ethnic background of the population.9 The prevalence of pSLE is consistently higher in girls than boys: Canadian and Taiwan studies estimated a factor of 6.12,13 Female predominance increases with age. This observation strongly supports the suggested role of female hormones.14 Although pSLE is less common than adult SLE, childhood onset was recently found to be a strong, independent predictor of overall lupus mortality.15
Children with pSLE are likely to have relatives with SLE. The pattern of familial aggregation for siblings with SLE suggests a polygenic inheritance.16 In addition, pSLE patients often have asymptomatic relatives with evidence of autoantibodies.17
Causes
Genes
Enabled by large international collaborations, susceptibility genes have been identified suggesting a dysregulated immune phenotype in lupus patients partially overlapping with other autoimmune diseases.18 These include genetic variants in the cytokine interferon-α pathway and their functional impact,19 the contribution of signal transduction STAT4 gene variations on lupus susceptibility,20 and the association of the interleukin-1 receptor-associated kinase-1 (IRAK1), an X chromosome gene, and disease susceptibility in pSLE.21
Beyond the type of gene variant (mutation, polymorphisms), altered copy number variations reflecting “gene dose” and epigenetic modifications of key lupus genes are reported in pediatric lupus patients: Garcia-Ortiz demonstrated an association of the lupus gene Toll-like receptor 7 (TLR7) copy number variation with susceptibility to pSLE in Mexican populations.22 Drug-induced SLE and incomplete monozygotic twin concordance rates suggest an important role of epigenetic factors such as histone modification or altered DNA methylation in pSLE.23 Finally, mitochondrial DNA polymorphisms may also be important in the pathogenesis of SLE.24 Children with inherited complement deficiencies including C2, C4A/C4B, C1q, and C1s can present with pSLE.25 These single gene defects are more commonly seen in familial cases of lupus.
Environment
Beyond genes and epigenetics, other potential contributing factors have been intensely studied in pSLE including environmental factors such as parental smoking26 and organic dust exposure.27 As a link between genetic and environmental factors, endogenous and external viruses are currently being studied in pSLE.28 The presence of an interferon signature pattern had raised suspicion for a potential viral contribution and discovery of the genetic basis of chilblain lupus.29 This focused the researchers’ attention to DNA repair of endogenous virus as a potential pathogenetic factor in pSLE.
Pathology
Similar to adult SLE, the inflammatory processes leading to organ dysfunction are heterogeneous between and within organs (see Chapter 79). Immunoglobulin deposition in small vessels such as glomerulus or lung capillaries, complement activation, antibody-binding to single cells, and microthrombotic or macrothrombotic vessel disease are the hallmarks of lupus. Hematologic manifestations of lupus are often related to direct antibody-binding and complement activation.30 In contrast, the histology of pediatric lupus nephritis is more variable and includes the distinct subtypes of mesangial, focal, or diffuse proliferative and membranous nephritis, which can coexist.31 Renal biopsies are required to distinguish subtypes and define treatment regimens. In children, severity of the glomerulonephritis on renal biopsy was shown to be associated with treatment choice and response and long-term outcome.31–34 Confounding risk factors for severe lupus nephritis and adverse renal outcome include evidence of thrombotic microangiopathy, antiphospholid antibodies, tubulointerstitial disease, hypertension, nephrotic syndrome, and access to health care.31–35
Clinical Features
The clinical presentation of pSLE was reported in large series from many countries, allowing a better understanding of the clinical diversity, impact of ethnicity, confounding factors including infections, and access to health care8,11,36–47 (Table 108-1).
Table 108-1 Clinical Characteristics of Children at Diagnosis of Pediatric Systemic Lupus Erythematosus (pSLE): Comparison of Four Recent Cohorts
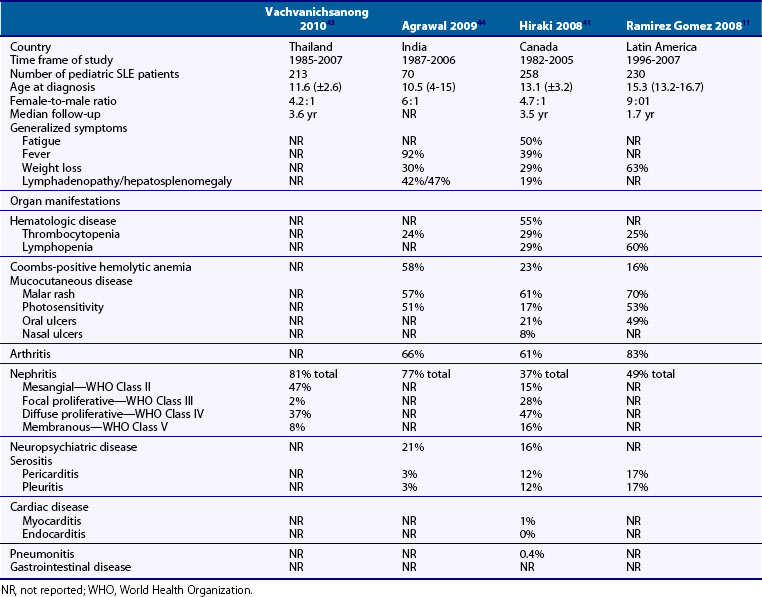
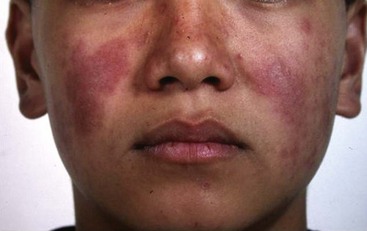
Systemic features including fever and fatigue are found in more than 90% of children at diagnosis of pSLE. Arthritis is the most frequently reported organ manifestation in pSLE. Typically pSLE arthritis is a nonerosive, painful polyarthritis. Mucocutaneous involvement includes the typical “butterfly” or malar rash, an erythematous rash in the malar distribution sparing the nasolabial folds (Figure 108-1). Diffuse hair loss is commonly seen in children with active disease. In addition, children with lupus can present with a photosensitive rash, exacerbated in sun-exposed areas including upper arms, neckline, and face; Raynaud’s phenomenon of fingers and toes; a vasculitic skin rash, which is often raised and painful affecting the fingers and toes; and oral and/or nasal ulcers. Oral ulcers are typically located on the hard palate and are painless (Figure 108-2). Uncommon skin manifestations include discoid lupus lesions, which heal with scarring.48
Nephritis is the most common major organ manifestation and occurs in more than 50% of children with pSLE, most commonly at diagnosis. Children with lupus nephritis present with peripheral edema, proteinuria, active urine sediment, and hypertension. Focal or diffuse proliferative glomerulonephritis accounts for more than 50% of all pediatric lupus nephritis in most series. Acute renal failure can occur in a third of children presenting with proliferative lupus nephritis.33 Mesangial and membranous nephritis are less common and can occur in conjunction with proliferative lupus nephritis. Posterior reversible encephalopathy (PRES) is an increasingly recognized central nervous system (CNS) complication of lupus nephritis and hypertension.49
Neuropsychiatric disease affects about a quarter of children with pSLE. Headaches in conjunction with psychosis or cerebrovascular disease presenting as seizures or severe cognitive dysfunction are the most common clinical phenotypes. Isolated lupus headache is uncommon.7 Psychosis in pSLE is characterized by optic and acoustic hallucinations and visual distortions. Many patients have overlapping features of cognitive dysfunction, headaches, and mood disorder.5 Isolated mood disorders such as depression are uncommon in pSLE. CNS vasculitis in pSLE more commonly affects the small vessels. Angiography-positive disease and strokes are uncommon.7 Transverse myelitis is an uncommon, serious manifestation of pSLE.50 Peripheral neuropathies are uncommon in children with lupus.
The diagnosis of NPSLE in children is based on clinical assessment including comprehensive neurocognitive testing,51 inflammatory markers, and neuroimaging. Children with neuropsychiatric disease commonly have antiphospholipid antibodies, in particular anti–β2-glycoprotein I (anti-β2GPI).52 A positive lupus anticoagulant is often detectable in children with cerebrovascular disease including sinus vein thrombosis (SVT) and chorea. Neuroimaging demonstrates only subtle abnormalities in half of patients5 including the majority of children presenting with psychosis.
Hematologic disease is common in pSLE and affects a quarter of children. Treatment-refractory idiopathic thrombocytopenic purpura (ITP) or severe autoimmune hemolytic anemia can be the presenting features of pSLE. A positive ANA and older age were found to be risk factors of pSLE in ITP.53 Serositis including pleuritis, pericarditis, and less commonly peritonitis affects about 20% of children with pSLE. Chest pain is the most common presenting feature. Inflammatory lung lesions including capillaritis and alveolar hemorrhage are uncommon. Although cardiomegaly due to pericarditis and arrhythmia/conduction anomaly occur frequently, myocarditis and coronary arteritis are serious yet uncommon lupus features in children.54,55 The most common gastrointestinal (GI) manifestation of pSLE is lupus hepatitis. Inflammatory bowel manifestations are rare. Overall atypical presentations can be found in up to a quarter of children ultimately diagnosed with pSLE and, when present, were found to correlate with poor outcome.56 Endocrinopathies of pSLE include hypothyroidism or hyperthyroidism and diabetes mellitus.57 Menstrual cycle disturbances and transient amenorrhea are common in girls with pSLE and may be associated with pituitary dysfunction or treatment with cyclophosphamide, leading to a decreased progesterone production.58
Diagnosis and Diagnostic Tests
The diagnosis of pSLE is based on clinical findings, laboratory test results including inflammatory markers, complement levels, markers of organ involvement, and specific autoantibodies. Tissue biopsies and imaging studies can further support and/or classify pSLE subtypes. The presence of 4 of 11 ACR classification criteria was found to have a sensitivity of 96% and a specificity of 100%.2 A careful evaluation of potential organ involvement of pSLE is mandatory. Characteristic abnormal laboratory markers of active pSLE may include a raised erythrocyte sedimentation rate (ESR), anemia, which may be Coombs positive and hemolytic, a low white blood count with predominant lymphopenia, low platelets, and low C3 and/or C4 complement levels. Paradoxically, the C-reactive protein (CRP) is normal in the vast majority of children with active lupus, except for those presenting with serositis or concurrent infections. An abnormal urinalysis including microscopic evidence of casts indicated renal involvement. Renal function impairment is best evaluated by serum creatinine, albumin, and urine protein–to-creatinine ratio. Lupus anticoagulant and specific lupus autoantibody testing including ANA, dsDNA, ENA, and antiphospholipid antibodies is mandatory. ANA is found is almost every child with pSLE, while dsDNA is detected in more than 80%.41,59 Novel antibodies have been proposed and require prospective validation in pSLE.60 Children may have frank hypothyroidism or hyperthyroidism or solely raised titers of thyroid antibodies.41
Lipid abnormalities are increasingly recognized in children with pSLE.61 Dyslipidemia may contribute to a risk of premature arteriosclerosis in pSLE patients.62,63 Cytokines derived from adipocytes including leptin, adiponectin, and ghrelin were recently shown to correlate with disease activity in pSLE.64 Markers of bone health and fracture risk have been extensively evaluated in children with pSLE.65 A comprehensive assessment may include these markers in conjunction with bone density measurement.
The differential diagnosis of pSLE is wide and includes infections such as cytomegalovirus (CMV), Epstein-Barr virus, and tuberculosis, malignancies such as lymphomas, endocrinopathies, primary inflammatory or idiopathic organ diseases such as membranoproliferative nephritis, or idiopathic psychosis and pediatric autoimmune diseases including JDM, systemic sclerosis, Sjögren’s syndrome, overlap syndromes, and polyarticular and systemic juvenile idiopathic arthritis.66,67 Children with a recent diagnosis of pSLE can present with concurrent infections such as CMV, which may confound the clinical presentation and response to therapy.68 Uncommon infections may be present.69 Macrophage activation syndrome (MAS, a secondary hemophagocytic lymphohistiocytosis [HLH] syndrome) is a newly recognized inflammatory emergency and can present in pSLE patients.70 MAS has to be considered in a child with pSLE and unexplained fever and cytopenia, when associated with marked hyperferritinemia.
Treatment
Immunosuppression
The choice and dosing of immunosuppressive therapy regimen must be tailored to the extent and severity of the child’s organ disease. A thorough diagnostic evaluation is mandatory before initiating therapy. Immunosuppressive treatment protocols are commonly adopted from adult trials and meta-analyses.71 Treatment response criteria for pSLE were recently developed and validated.72
Corticosteroids are the mainstay of lupus therapy. The general approach for major organ disease includes initial high-dose treatment with 2 mg/kg prednisone equivalent in two to three divided doses followed by a slow taper. This includes treatment of proliferative lupus nephritis, neuropsychiatric lupus except for chorea and SVT, myocarditis, and lung disease. Pulse intravenous (IV) methylprednisolone is frequently used for emergent situations including acute psychosis, MAS, and myocarditis. Arthritis, serositis, nonproliferative lupus nephritis, and mucocutaneous disease may require smaller initial doses of corticosteroids. The efficacy of corticosteroids in pSLE is well established. However, the significant toxicity limits its long-term use at high doses. Short-term side effects include weight gain, sleep disturbances, emotional instability, increased hair growth, and impaired glucose metabolism. Long-term effects include cataracts, growth arrest, vertebral fractures, and avascular necrosis.57
Combination immunosuppressive regimens are commonly used in children with major organ involvement. Cyclophosphamide, mycophenolate mofetil (MMF), and azathioprine have been studied in observational cohorts of pSLE patients. IV cyclophosphamide was considered the gold standard for severe organ disease such as proliferative lupus nephritis and neuropsychiatric disease. In accordance to adult SLE protocols, cyclophosphamide is commonly used for induction over 6 months, followed by either azathioprine or MMF.73 Induction therapy with MMF was found to be safe, well tolerated, and effective in a small cohort of renal and nonrenal pSLE patients.74–77 Complete renal remission is achieved in 40% to 50% of children at 6 months and 75% at 12 months.78 Commonly reported side effects that led to discontinuation of MMF therapy included severe diarrhea and abdominal pain.75 In children, optimal dosing may require pharmacokinetic evaluation on a stable dose.79,80 Efficacy and safety of MMF as maintenance drug is well established in pSLE.73,81,82 Induction therapy with azathioprine was found to be equally efficacious as cyclophosphamide in children with proliferative lupus nephritis and renal failure.33 Azathioprine has a good efficacy and safety profile.83 Routine monitoring of blood counts and liver function is required. Significant toxicity may occur in children with mutations in the gene encoding thiopurine methyltransferase or thiopurine S-methyltransferase (TPMT); however, the role of genotyping remains controversial.84 Azathioprine is also used as a maintenance drug following induction with cyclophosphamide.73
Dialysis is required in children with end-stage renal disease.33 Plasmapheresis is indicated for specific disease manifestations such as thrombotic-thrombocytopenic purpura (TTP),85 transverse myelitis, and steroid-resistant nephritis.86 B cell depletion with the anti-CD20 antibody rituximab is the main biologic therapy currently used in pSLE.87–91 It was shown to be effective as a single agent in hematologic disease and in addition to standard therapy in refractory, difficult-to-treat pSLE. The safety profile remains to be systematically studied in pSLE. Autologous stem cell transplantation is rarely performed in pSLE.92
MAS therapy in pSLE may include IV immunoglobulin, IV methylprednisolone pulse therapy, cyclosporine, or even chemotherapy according to HLH protocol.93
Antimalarials
Antimalarials are strongly recommended for children and adults with SLE.94 On the basis of predominantly adult studies and meta-analysis, antimalarials are thought to decrease overall mortality and improve long-term outcome,95 modify lipid profiles,96 and control joint and skin disease, in particular discoid lupus lesions.97,98
Adjunctive Therapy
Vitamin D is known to be a strong factor of bone protection in pSLE.99,100 Sufficient vitamin D doses in addition to calcium intake and physical activity are required to maintain good bone health. More recently a novel role of vitamin D in maintaining immune homeostasis was recognized. This is supported by studies demonstrating an inverse correlation between vitamin D levels and disease activity,101,102 in particular in overweight children with pSLE.
Outcome
Lupus in children and adults is a relapsing/remitting disease. The burden of pSLE is complex to determine because many factors such as access to health care, individual patient characteristics, disease activity, confounding diseases such as infections, and responsiveness to treatment all contribute to overall mortality and morbidity.1
The overall mortality as captured by standard mortality rate (SMR) for all SLE in the United States between 1992 and 2001 was 3.06 deaths per million inhabitants per year,103 in Brazil between 1985 and 2004 it was 3.8 (2601 deaths, 90% female),104 and in Denmark it was 4.6.105 With improved therapies, mortality rates were shown to decrease in Canada (SMR, 10.1 in 1970-1977; 4.8 in 1978-1985, and 3.3 in 1986-1994).106 When comparing childhood- with adult-onset SLE, childhood-onset SLE was found to be independently associated with an increased mortality risk (hazard ratio [HR], 3.1), as was low socioeconomic status measured by education (HR, 1.9), and end-stage renal disease (HR, 2.1).15
Young age at disease onset was repeatedly shown to be a predictor of adverse outcome.47,107 Children with pSLE in poor countries clearly have a higher mortality: In a small study from Nigeria the mortality was 30%.108 The Latin American LUMINA cohort had an 81% survival at last follow-up, the recent 5-year patient survival rate in Iran was 82.5%,32 and in Canada it was 100%.41,109 Infections continue to be the main cause of death in developing countries with limited access to health care.34,110 Nephritis has been consistently identified as a predictor of poor outcome in pSLE. Histologic subtype of proliferative disease, evidence of disease relapse, certain ethnicities, and poor response to therapy were strong predictors of end-stage renal disease in pSLE.111 Gibson demonstrated that treatment resistance portended a high risk of end-stage kidney disease and disproportionately affected African-American children with lupus nephritis.111
Children with pSLE accrue disease- and treatment-related damage as captured in the domains of the Systemic Lupus International Collaborative Clinics (SLICC) Damage Index (see Chapter 80), constantly adding to the overall disease burden.1,57 Osteoporosis, cataracts, and osteonecrosis/avascular necrosis (AVN) are the leading domains of damage accrual. Individual patient characteristics, disease activity, corticosteroid therapy, calcium/vitamin D deficiency, and immobility contribute to impaired bone health in pSLE.112,113 AVN occurs in 6% to 10% of pSLE patients overall and is associated with corticosteroid therapy.41 Nakamura114 recently observed the complete absence of AVN in children younger than 14 years of age and suggested that age at the time of the initial corticosteroid therapy affects AVN occurrence. Neurocognitive deficits secondary to disease and treatment are increasingly recognized and significantly affect school performance and overall health-related quality of life.115,116 Early cardiovascular events including myocardial infarctions and strokes have become a major cause of morbidity and mortality.1
Drug-Induced Lupus Erythematosus
Several medications can cause systemic and subacute or chronic cutaneous lupus phenotypes in children.117 The cutaneous manifestations of systemic drug-induced lupus (DIL) include malar rash, purpura, erythema nodosum, urticaria, and photosensitivity. Systemic symptoms include arthritis, oral ulcers, pleuritis, hematologic manifestations, and less commonly renal disease. Characteristic laboratory findings of DIL are positive ANA and antihistone antibodies. Drugs implicated are minocycline, anticonvulsive drugs, hydralazine, procainamide, and isoniazid.118
Neonatal Lupus Erythematosus
Neonatal lupus erythematosus (NLE) is an acquired disease of the newborn caused by placental transfer of maternal anti-SSA/Ro and anti-SSB/La IgG antibodies. These can be present in mothers with SLE, Sjögren’s syndrome, and other autoimmune connective disorders, as well as clinically healthy women. Antibody transfer can lead to inflammation of the cardiac conducting system and subsequent fibrosis resulting in congenital heart block (CHB), which may be detected as early as 20 weeks of gestation. A prolonged PR interval is the first electrocardiographic sign of conduction system abnormality in NLE. The degree of heart block can vary, and rapid clinical progression from normal sinus rhythm to complete CHB over 2 weeks may be observed, causing life-threatening cardiomyopathy and fetal hydrops in the most severe cases. Isolated endocardial fibroelastosis can be found in some infants.119 Interestingly, infants with prenatal exposure to high-titer anti-SSB/La antibody levels are more likely to have noncardiac features of NLE, whereas cardiac disease tends to be associated with moderate or high maternal anti-SSA/Ro levels, independent of anti-SSB/La titers in CHB.120 The overall risk of CHB in anti-SSA/Ro–positive women is estimated to be 2% to 5%,120 but this risk may be increased by 10-fold in women with a previous child with CHB.121 A recent study suggests an overall recurrence rate of cardiac NLE of 17%, independent of maternal health, antenatal use of steroids, antibody status, severity of cardiac disease in the first affected child, or sex of the subsequent child.122
In addition to heart block, newborns with NLE can present with a characteristic NLE rash, hepatic dysfunction, and hematologic abnormalities including significant thrombocytopenia. Typically the NLE rash is located around the eyes but may present elsewhere on the body.123 Hepatobiliary disease can have three distinct presentations: (1) transient conjugated hyperbilirubinemia with mildly raised liver function tests (LFTs) in the first weeks of life; (2) mild elevations of LFTs at 2 to 3 months of life; and (3) severe liver failure during gestation or in the neonatal period.124 NLE neurologic involvement can include magnetic resonance imaging (MRI) findings of nonspecific white matter changes and calcification of the basal ganglia. NLE “vasculopathy” is reported. Recently, an association of NLE and hydrocephalus has been recognized.125 Chondrodysplasia punctata, a stippling of the epiphyses, and pulmonary capillaritis are rare clinical presentations of NLE.123,126
In a prospective multicenter study of 128 infants whose mothers had been referred for the presence of anti-SSA/Ro antibodies, regardless of their diagnosis, hematologic abnormalities and raised liver enzymes were seen in 27% and 26%, respectively.127 Cutaneous NLE manifestations were present in 16%. Only 2 of the 128 infants (1.6%) presented with complete CHB. In a recent Japanese review, 193 infants with NLE were described reporting CHB in 23%.128
Treatment of CHB in NLE remains controversial. Prevention of progression to complete CHB may be achieved by treating the mother with fluorinated steroids (dexamethasone or betamethasone), which are not metabolized by the placenta and are available to the fetus in an active form. IV immunoglobulin had been used to prevent the development of CHB in the index patient and in subsequent pregnancies.129 The current recommendation is to screen anti-Ro/SSA antibody–positive mothers with serial echocardiograms and obstetric sonograms biweekly starting from week 16 of gestation. Early detection of cardiac manifestations of NLE including premature atrial contractions or moderate pericardial effusion preceding CHB may potentially be targeted with preventive therapy.129–131 First-degree heart blocks can be reversed by dexamethasone treatment of the mother.132 Once third-degree block is unequivocally identified, reversal is unlikely to be achieved. The majority of children with CHB require pacemakers.121
The treatment approach to extracardiac manifestations of NLE is conservative. Skin disease may require topical corticosteroids and sun protection.133 Transient elevations of LFTs and cytopenias commonly do not require therapy.124
The morbidity of cardiac neonatal lupus is estimated to be 20%.121 Mortality is particularly high in patients with CHB and concurrent cardiomyopathy. Children with NLE can develop SLE later in life. Concerns of potential long-term neurocognitive deficits of NLE patients need further evaluation.134
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


