98 Pathogenesis of Osteoarthritis
Expression of inducible cyclooxygenase-2 is increased in osteoarthritis chondrocytes.
Most current treatments aim to improve the signs and symptoms of osteoarthritis.
In industrialized societies OA is the leading cause of physical disability, increases in health care usage, and impaired quality of life. The impact of arthritic conditions is expected to grow as the population both increases and ages in the coming decades.1 Despite its prevalence, the precise etiology, pathogenesis, and progression of OA remain beyond our understanding, primarily due to multiple confounding factors. Without a clear-cut picture of how OA arises at the cellular or molecular level, many still consider it a result of “wear and tear,” an inevitable consequence of aging. In fact, the accompanying biochemical, structural, and metabolic changes in joint cartilage have been well documented. It is now known, for example, that cytokines, mechanical trauma, and altered genetics are involved in its pathogenesis and that these factors can initiate a degradative cascade that results in many of the characteristic alterations of articular cartilage in OA. More recently it has become apparent that OA is a disease process that affects the entire joint structure including cartilage, synovial membrane, subchondral bone, ligaments, and periarticular muscles. OA is thus better considered a group of overlapping disorders of various etiologies arising from a combination of systemic factors (e.g., genetics) and local factors (e.g., biomechanically or biochemically mediated events) that gradually converge to produce a condition with definable morphologic and clinical outcomes.2
Etiologic Factors in Osteoarthritis
Age
Age is the risk factor most strongly correlated with OA.3,4 OA is the most common chronic disease in later life; more than 80% of persons older than age 75 years are affected, and OA increases progressively with age at all joint sites. Radiologic changes in OA increase as individuals age,5 although these changes do not always correlate with clinical symptoms or disability.6,7 Although clearly an age-related disease, OA is not, however, an inevitable consequence of aging. Age-related morphologic and structural changes in articular cartilage include fraying, softening, and thinning of the articular surface; decreased size and aggregation of matrix proteoglycans; and loss of matrix tensile strength and stiffness. These age-related tissue changes are most likely due to a decrease in the chondrocytes’ ability to maintain and repair the tissue, as chondrocytes themselves undergo age-related decreases in mitotic and synthetic activity, exhibit decreased responsiveness to anabolic growth factors, and synthesize smaller and less uniform large aggregating proteoglycans and fewer functional link proteins.4
There also appears to be a direct correlation between chondrocyte apoptosis and cartilage degradation leading to OA. Age does appear to be an independent factor that predisposes articular chondrocytes to apoptosis because the expression levels of specific proapoptotic genes (Fas, FasL, caspase-8, and p53) is higher in aged cartilage.8,9
Joint Location
Although osteoarthritis occurs most commonly in weight-bearing joints,10 age affects joints differentially.11 A study comparing tensile fracture stress of cartilage in the femoral head and in the talus showed that it decreased progressively with age in the former but not in the latter.12 Joint-specific age-related viability in articular cartilage may explain why OA is more common in hip and knee joints with increasing age but occurs rarely in the ankle.
Obesity
Obesity is another important risk factor for OA.13–15 Greater body mass index in both women and men has been shown to be associated with an increased risk of knee (but not hip) OA.7,16,17 An increase in mechanical forces across weight-bearing joints is probably the primary factor leading to joint degeneration. Obesity not only increases the forces at weight-bearing joints but may also change posture, gait, and physical activity level, any or all of which may further contribute to altered joint biomechanics.18 The majority of obese patients exhibit varus knee deformities, which results in increased joint reactive forces in the medial compartment of the knee, thereby accelerating the degenerative process.19
Particularly in elderly obese individuals, heavy physical activity is an additional risk factor for the development of knee OA, whereas light to moderate activity does not appear to increase risk for knee OA and may in fact alleviate symptomatic knee OA by reducing body mass index.7,16,17 Similarly, weight loss can reduce both radiographic knee OA progression and clinical symptoms. Recent evidence indicates that in obese patients with OA, significant weight loss dramatically improves functional status, with short-term results equivalent to those of patients who have undergone joint replacement.20
Yet recent work suggests that the association between obesity and osteoarthritis lies beyond the mechanical loading from a higher body mass index. Adipose tissue is now recognized as a metabolically active contributor to inflammatory cascades. Activated adipose tissue increases the synthesis of proinflammatoy cytokines including leptin, adiponectin, resistin, interleukin-1 (IL-1), IL-6, and tumor necrosis factor (TNF), while some regulatory cytokine levels such as IL-10 are decreased.21,22 Specifically, the discovery of the obesity gene and its product leptin may have important implications for the onset and progression of OA and increase our understanding of the link between obesity and OA. The fact that women have a greater proportion of total body fat and relatively higher levels of adipose-derived systemic leptin concentrations than men may partially account for the gender disparity in OA patients. Leptin, however, is produced not only by adipose cells but also by osteoblasts and chondrocytes, suggesting that local leptin production may play a role in OA. Significant levels of leptin were observed in the cartilage and osteophytes of subjects with OA, yet few chondrocytes produced leptin in the cartilage of healthy subjects. Leptin has also been demonstrated to induce anabolic activity in the chondrocytes of rats and thus may ultimately confer structural joint changes.23,24
Genetic Predisposition
Because of the prevalence of OA in the general population and its extensive clinical heterogeneity, the genetic contribution to its pathogenesis has been difficult to analyze.25,26 Two major cohorts (the Framingham Study and the Baltimore Longitudinal Study on Aging) support a genetic contribution to OA, with evidence for a major recessive gene and a multifactorial component, representing either polygenic or environmental factors.27,28 Twin pair and family risk studies have indicated that the heritable component of OA may be on the order of 50% to 65%.26,29,30 Moreover, family, twin, and population studies have indicated differences among genetic influences that determine the site of OA (hip, spinal, knee, hand).28,31,32 Further evidence supporting a genetic predisposition to OA is the demonstration of a significantly higher concordance for OA between monozygotic twins than between dizygotic twins. Genetic studies have identified multiple gene variations associated with an increased risk of OA.33
Inherited forms of OA may be caused by mutations in several genes that are expressed in cartilage including those encoding types II, IV, V, and VI collagens, as well as cartilage oligomeric matrix protein (COMP).34,35 Recently it was reported that mice deficient in the type IX collagen gene and the matrilin 3 gene (human equivalent condition not yet reported) developed age-dependent OA-like changes in the knee and temporal mandibular joints.36,37 Moreover, candidate genes in OA have been identified that are not structural proteins. The haplotype of a vitamin D receptor (VDR) that plays a vital role in controlling bone mineral density appears to be associated with a twofold risk of knee OA38–40—although the VDR locus is close to the COL2A1 locus on chromosome 12q, so the association may be due to linkage disequilibrium with the latter.25 In addition, the locus of the insulin-like growth factor 1 (IGF-1) gene has been associated with radiographic OA, as has an aggrecan polymorphic allele with hand OA.25
In population studies, genome-wide linkage scans have highlighted up to seven chromosomal regions that may harbor OA susceptibility genes.41 Chromosome 2q was positive in several scans, suggesting that this chromosome is likely to harbor one or more susceptibility genes. In a study of affected sibling pairs, a region of linkage stretching from 2q12 to 2q21 was reported for OA of the distal interphalangeal joint and a previous study of affected sibling pairs in the United Kingdom demonstrated a broader region of linkage, stretching from 2q12 to 2q31.41,42 Two IL-1 genes (IL1A and IL1B) and the gene encoding IL-1Ra (IL1RN) are located on chromosome 2q13 within a 430-kb genomic fragment. Given the importance of IL-1 in the perpetuation of cartilage damage in OA, it is possible that a proportion of the genetic susceptibility to OA may be encoded for by variation in the activity of interleukins, and that for chromosome 2q this susceptibility could reside within the IL-1 gene clusters. Loughlin and colleagues,41 however, have provided evidence that the IL-1 gene cluster harbors susceptibility for knee OA, but not for hip OA. These and other epidemiologic studies have highlighted potential differences in the degree of OA heritability among different joint groups and between the two sexes.43,44
Genomic and postgenomic technology, in addition to defining susceptibility genotypes, will also lead to the discovery of genes and gene products that are overexpressed in OA tissues and that contribute to disease pathogenesis and progression.45–47 Studies of differential gene expression in diseased tissue, in addition to elucidating pathogenic processes that lead to novel therapies, could also have two additional benefits: (1) identification of unique biomarkers that can be used for OA diagnosis or management and (2) identification of candidate susceptibility genotypes such as polymorphic variations of cytokines or growth factors that may predispose to disease progression.48
Joint Malalignment and Trauma
Joint malalignment or trauma may lead to rapid development of OA, or it may initiate a slow process that results in symptomatic OA years later. Probably as a result of progressive reduction in periarticular blood flow and the resultant decrease in rate of remodeling at the osteochondral junction, joints become increasingly congruent with age.49,50 Altered joint geometry may interfere with nutrition of the cartilage, or it may alter load distribution, either or which may result in altered biochemical composition of the cartilage, irrespective of age.51,52 Local factors such as stresses related to joint use and joint deformity also influence the development of OA.
Joint incongruence (e.g., malreduced intra-articular fractures, developmental dysplasia of the hip, recurrent dislocation of the patella) can lead to early-onset OA.53 Repetitive, high-impact sports are strongly associated with joint injury and increase the risk for lower limb OA.54 Repetitive trauma at a subfracture level has been shown to accelerate remodeling in the zone of calcified cartilage, with reduplication of the tidemark and thinning of the noncalcified zone, resulting in stiffening of the subchondral bone, increased wear of the overlying cartilage, and ultimately development of OA.55–57 Regular exercise is important in maintaining articular cartilage structure and metabolic function. Recreational running and low-impact activities have not been shown to increase the risk of OA.
Articular cartilage is remarkably resistant to damage by shear forces; it is, however, highly vulnerable to repetitive impact loading.58 When joints are subjected to in vitro cyclic loads that are easily borne by subchondral bone, cartilage degeneration still results.59 This vulnerability accounts for the high frequency of OA in shoulders and elbows of pneumatic drill operators and baseball pitchers, ankles of ballet dancers, metacarpophalangeal joints of boxers, and knees of basketball players. The risk for knee osteoarthritis among participants in sports, however, may be more closely related to previous knee injury than to participation in sport alone.60
The major forces on articular cartilage, in addition to weight bearing, are due to the contraction of the muscles that stabilize or move the joint.61 For example, in normal walking, 4 to 5 times the body weight may be transmitted through the knee, and in squatting it is up to 10 times the body weight.39 Articular cartilage is believed to be too thin to be an effective shock absorber under these high loads. What protects the joint under physiologic conditions of impact loading is joint motion, with the associated lengthening of muscles under tension and deformation of the subchondral bone.40,62 Cancellous subchondral bone functions as a major shock absorber due to its material properties.56 Two-thirds of subchondral bone stiffness derives from bony trabeculae, about one-third from intraosseous fluid.63 In the normal unloaded joint, the opposing surfaces are not congruous; under loading, both the cartilage and the bone deform so that a larger proportion of the opposing surfaces comes into contact, increasing joint congruity and resulting in a force distribution over the largest possible area.64 Excessive loads may cause microfractures of subchondral trabeculae that heal via callus formation and remodeling, resulting in stiffer-than-normal bone that is less effective as a shock absorber and predisposing articular cartilage to degeneration.
Whether subchondral sclerosis precedes the onset of OA or is a change secondary to cartilage degeneration is not known. Indirect evidence supports the theory that biomechanical changes in subchondral bone may be important in OA.63,65,66 Foss, for example, reported cases of femoral osteoporosis (which is associated with softening and greater compliance of the subchondral bone) that may have protected the hip from OA.67 Conversely, in vitro studies have shown that stiffening of the cancellous bone with methacrylate, reducing its deformability, leads to cartilage degeneration with repetitive impact loading.68
Gender
Women are about twice as likely as men to develop OA. Although women have a lower prevalence of OA than men before age 50 years, there is a marked increase in prevalence among women after 50, particularly in the knee.69 Radiographic and interview data from the National Health and Nutrition Examination Survey (NHANES III), a representative cross-sectional health examination survey of the U.S. population, reported that the lifetime prevalence of radiographic knee osteoarthritis was 37.4% and that the prevalence of symptomatic knee osteoarthritis was 12.1% in adults age 60 years and older; prevalence was greater among women than men (42.1% vs. 31.2%), and women had significantly more Kellgren-Lawrence grade 3 and 4 changes (12.9% vs. 6.5% in men).70 Women have a greater number of joints involved and are more likely to exhibit clinical symptoms of morning stiffness, joint swelling, and nocturnal pain. The gender differences in OA incidence after age 50 may be the result of postmenopausal estrogen deficiency. Articular chondrocytes possess functional estrogen receptors, suggesting that these cells can be regulated by estrogen. Nuclear estrogen receptors (ERs) have been detected in articular chondrocytes of humans,71,72 rats,72,73 monkeys,72,74 pigs,75 and human growth plate chondrocytes.76
Recent epidemiologic studies have linked estrogen replacement therapy (ERT) with a lower-than-expected risk of knee and hip OA in postmenopausal women. Clinical investigations of the association between OA and hormonal level in women have involved measurement of circulating estrogen levels in postmenopausal women, general radiographic evaluation of postmenopausal women, and examination of the effect of ERT on such variables as knee OA and cartilage volume.77–80 In a study of more than 4000 women age 65 years or older that assessed pelvis radiographs for hip OA, Nevitt and colleagues77 showed that women using oral estrogen faced a significantly reduced risk of hip OA. Estrogen users for 10 years or longer had a greater reduction in risk of developing hip OA than users for less than 10 years. Zhang and colleagues,79 using weight-bearing radiographs in female participants in the Framingham Osteoarthritis Study (n = 831; mean age, 73 years; age range, 63 to 93) to study the rate of knee OA, reported a modest but nonsignificant greater protective effect for radiographically detected OA in women who were on ERT. A prospective cohort study using anteroposterior weight-bearing knee radiographs of women (mean age, 71 years; age range, 63 to 91) from the Framingham Study categorized patients into three groups according to estrogen use at biennial examination: never users (n = 349), past users (n = 162), and current users (n = 40). When both incident and progressive radiographic knee OA cases were combined, current ERT users were found to have 60% less risk for knee OA than never users. Wluka and colleagues80 reported on the longer-term use of ERT and its association with knee cartilage volume (measured by magnetic resonance imaging [MRI]) in postmenopausal women; results showed that after adjusting for confounders, women using long-term ERT had more knee cartilage than controls. Beneficial effects of ERT on the severity of knee OA in ovariectomized monkeys have also been demonstrated.81
Changes in Osteoarthritis
Early Reparative, Proliferative, and Hypertrophic Changes
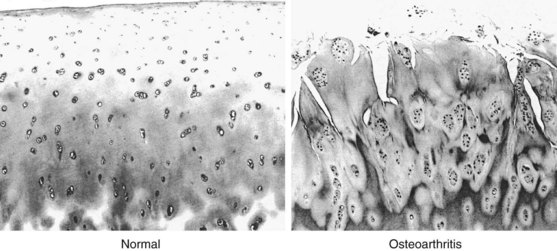
Attempts at local self-repair can be seen as an initial increase in the number of chondrocytes in the form of clusters or clones, with as many as 50 or more cells in a cluster39 (Figure 98-1). In healthy cartilage, chondrocytes are quiescent and neither commit to proliferation nor to further hypertrophic differentiation. In contrast, in the early stages of OA, proliferating chondrocytes in clusters express higher levels of matrix proteins including aggrecan and type II collagen,83 as well as stem cell markers84 and markers of hypertrophic differentiation.83 The chondrocyte clusters with proliferative and hypertrophic markers are a hallmark of OA and are also prominent in animal models of OA and three-dimensional cell cultures.85 In addition to being a repair response, chondrocyte clusters are thought to contribute to OA pathogenesis and progression through the release of matrix-degrading enzymes, growth factors, and inflammatory cytokines that affect surrounding chondrocytes and joint tissues.85
Osteophyte Formation
Osteophytes consist of newly formed fibrocartilage and bone and are most commonly formed at the peripheral margins of joints at the interface between cartilage and the periosteum. Osteophytes are thought to arise through chondrogenic differentiation of progenitor cells, most commonly from within the periosteum.86 As such, osteophytes may be a cellular repair response to the altered growth factor environment after joint injury, and in certain cases osteophytes can contribute to the stability of the joints.87,88 The connection between osteophyte formation and the repair response is further strengthened by animal models of OA in which the early stages of osteophyte formation can be histologically apparent as early as 3 days after joint injury. Many growth factors that enhance in vitro chondrogenic differentiation of stem cells are also present in osteophytes, including members of the transforming growth factor (TGF)-β superfamily,89 bone morphogenetic proteins (BMPs),90 IGF, and fibroblast growth factor,86 and TGF-β can induce osteophyte formation in animal models.91 As OA progresses, the osteophytes can limit movement and become painful.
Hypocellularity
A reduction in cell number is observed in aging cartilage, and the reduced synthetic capability of hypocellular cartilage is a contributing factor to the initiation and progression of OA. In healthy adult femoral cartilage, the cell density varies between 24,000/mm3 in the surface zone and 8000/mm3 in the deep zone, with an average of 1.65% of the cartilage volume consisting of cells.92 This number is greatly reduced in OA, through cell death via necrosis or apoptosis.93 Necrosis can result from direct mechanical damage to cells and is not generally an active process, whereas apoptosis is an active energy-consuming process. Apoptotic chondrocyte death can be initiated by many factors that are involved in the initiation and progression of arthritis including mechanical damage or injury, changes in cell-matrix interactions, oxidative stress resulting from nitric oxide or other reactive oxygen species, impaired mitochondrial function, and signal transduction pathways such as CD95/CD95 ligand. These pathways ultimately converge, and the execution of apoptotic cell death is mediated by the activation of caspases. Inhibition of apoptosis by interfering with caspase activation following injury is being explored as a chondroprotective intervention to prevent secondary OA following injury. Compromised regulation of cellular senescence and autophagy can also cause cell death. The role of autophagy in inflammation and pathogenesis has been gaining the attention of researchers.94 In some circumstances cell death can be mediated by the autophagic degradation of the damaged organelles. In cartilage, autophagy can be protective, and its reduction in OA corresponds with increased apoptotic markers.95
Alterations in Cartilage Matrix Metabolism
Biochemical Changes
These morphologic changes are accompanied by changes in the biochemical composition of the cartilage, which vary from early to later stages in the disease process. In early OA, the water content of the articular cartilage significantly increases, causing the tissue to swell and altering its biomechanical properties. This phenomenon suggests that there has been weakening of the collagen network; the type II collagen fibers have a smaller diameter than normal cartilage, and the normally tight weave in the midzone is slackened and distorted.96–101
In later stages of OA, type I collagen concentration within the extracellular matrix increases and the proteoglycan concentration falls to 50% or less than normal, with less aggregation and shorter glycosaminoglycan side chains.98,102 Keratan sulfate concentration decreases, and the ratio of chondroitin-4-sulfate to chondroitin-6-sulfate increases, reflecting synthesis by chondrocytes of a proteoglycan profile more typical of immature cartilage.103 Proteoglycan concentration in the cartilage diminishes progressively until the end stages, when histologic staining detects little or none.104
Our understanding of how the biochemical changes progress from the early to the later stages of OA is evolving. One of the first steps in cartilage degradation is a decrease in the density of proteoglycans. This step is thought to be at least partly reversible.105 However, decreased proteoglycan density opens up the cartilage porosity to make it more permeable to collagenases and other proteases, and it exposes collagen fibrils. This initiates a vicious circle of positive-feedback loops that further promote cartilage degradation. For example, epitopes on collagen become accessible to the DDR2 receptor on the cell surface, which then increases MMP-13 production through activation of the Ras/Raf/MEK/ERK and p38 signal cascades.106 The partially digested matrix components themselves have cytokine-like activity that enhances the inflammatory response and promotes matrix degradation. The destruction of the collagenous cartilage component is thought to be irreversible.
Calcium crystals (e.g., calcium pyrophosphate dihydrate, basic calcium phosphate crystals) are commonly found in the cartilage of the elderly, and often crystal arthropathy coexists with OA.107 That calcium crystals play a role in causing or worsening OA is supported by clinical and laboratory studies, but the relationship is complex and it is unclear whether these crystals are directly involved in OA pathogenesis.108–110 Pyrophosphate (PP) is produced from adenosine triphosphate (ATP) by the exoenzyme nucleoside pyrophosphohydrolase.111 High levels of PP in synovial fluid from OA patients correlate directly with the severity of joint damage.112,113 Young or proliferating chondrocytes are a major source of PP, whereas resting chondrocytes from normal adult cartilage secrete little.111 Thus the increased PP secretion in OA cartilage might indicate increased chondrocyte metabolic activity toward matrix repair.114 The presence of CPPD may alter the biomechanical properties of the cartilage extracellular matrix and lead to cartilage breakdown. Hemochromatosis (hemosiderin), Wilson’s disease (copper), ochronotic arthropathy (homogentisic acid polymers), gouty arthritis (crystals of monosodium urate), and calcium pyrophosphate dihydrate (CPPD) crystal deposition disease are further examples of conditions in which the abnormal entity may alter the cartilage extracellular matrix, leading to either direct or indirect chondrocyte injury by increasing the stiffness of the tissue and thereby precipitating the development of OA.
Metabolic Changes
Early OA is characterized by increased synthesis of proteoglycans, collagen, noncollagenous proteins, hyaluronate, and cell replication.104,115 This “activation” of chondrocytes is thought to be an attempt to repair the cartilage matrix, although it is not always effective and yields a matrix of inferior quality that is more susceptible to degradation.116 However, both anabolic and catabolic processes increase as cells attempt to repair or maintain tissue integrity98 and it is the imbalance between synthesis and degradation that is important in the pathogenesis of OA.117 In later stages of OA, there is a decrease in the synthesis of matrix per cell and a decrease in cell number. Furthermore, the quality of the synthesized matrix is reduced with respect to the composition and distribution of their glycosaminoglycans, the size of the proteoglycan subunit, and their ability to aggregate with hyaluronic acid.98,100,103,118,119 In addition to the reduced matrix production and hypocellularity, there is increased synthesis and activation of matrix degrading enzymes, as well as an overall decrease in the concentrations of enzyme inhibitors such as tissue inhibitors of metalloproteinases (TIMPs). Eventually OA progresses when the chondrocytic anabolic repair processes cannot keep pace with catabolic processes.98,104 The complex interaction between matrix synthesis and degradation explains why OA typically is slowly progressive and at times remains static by morphologic criteria, but ultimately it results in the overall degradation of cartilage matrix. The following sections summarize the major anabolic and catabolic factors relevant to cartilage degradation and the pathogenesis of OA.
Anabolic Factors (Transforming Growth Factor-β, Bone Morphogenetic Proteins) and Cartilage Repair
TGF-β is essential for the formation and maintenance of cartilage (reviewed in Reference 120). Interference with TGF-β function in cartilage leads to OA in multiple animal models121 and in human genetic susceptibility to OA.122 TGF-β affects cartilage homeostasis at multiple levels: It enhances stem cell chondrogenesis to increase the pool of cells available for cartilage synthesis, and it increases matrix production in existing chondrocytes. TGF-β also increases synthesis of anticatabolic factors such as TIMPs and PAI-1, proteins that inhibit the activation of latent proteinases in cartilage. TGF-β attenuates the cellular response to inflammatory cytokines including IL-1β and TNF,120 and TGF-β signaling through the Smad2/3 pathway can inhibit terminal differentiation and hypertrophy in chondrocytes. However, TGF-β activity can also contribute to the pathogenesis and progression of OA. For example, TGF-β signaling induces osteophyte formation in animal models. In aging cells and cells in which the ratio of the TGF-β receptors ALK1 and ALK5 is altered, TGF-β can also have opposing effects such as activating MMP-13 and inducing terminal hypertrophic differentiation in chondrocytes.123
BMPs are structurally related to TGF-β but generally activate a different set of receptors and intracellular signaling molecules. BMPs influence all stages of embryonic chondrogenesis and can promote chondrogenic differentiation of adult MSCs.124 Recent genetic evidence suggests that impaired BMP signaling affects OA susceptibility, with the most progress reported for BMP-14 (GDF-5).125,126 There is good evidence that a reduction of BMP-7 (OP-1) is evident in osteoarthritic cartilage,127 with possible regulation occurring through both inhibitory microRNA128 and promoter methylation.129 Supplementation of BMP-7 in joints reduces experimental arthritis in animals and was proven safe in a phase I clinical trial, with no findings of ectopic bone formation in the enrolled patients during the study.130 However, as described for TGF-β earlier, BMP signaling through certain receptors can also enhance terminal differentiation and hypertrophy in chondrocytes, processes that are hallmarks of OA progression.
Catabolic Factors and Cartilage Degradation
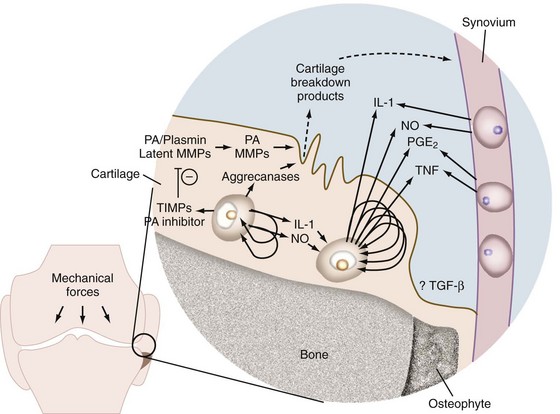
In addition to new matrix synthesis, cartilage remodeling also involves a degree of proteolysis. This occurs via the induction of an array of proteases, particularly matrix metalloproteinases (MMPs). In OA, the cytokines IL-1 and TNF stimulate the synthesis and secretion of many proteases and MMPs (Figure 98-2).105 IL-1 is synthesized by mononuclear cells (including synovial lining cells) in the inflamed joint and by chondrocytes as an autocrine activity.131–133 The enzymes stimulated by IL-1 and TNF include latent collagenase, latent stromelysin, latent gelatinase, aggrecanase, and tissue plasminogen activator (TPA).134–137 Plasminogen is either synthesized by the chondrocyte or enters the matrix by diffusion from the synovial fluid. TPA converts plasminogen to plasmin, a serine proteinase that can activate latent cartilage-degrading enzymes. A downstream mediator of IL-1- and TNF-induced cartilage degradation that is gaining increasing attention is hypoxia-inducible factor 2α (Hif2α).138,139 Hif2α is a transcription factor strongly upregulated in OA cartilage and mouse models of OA. It directly induces the expression of many cartilage-degrading enzymes including MMPs 1, 3, 9, and 12, as well as a disintegrin and metalloproteinase with thrombospondin motif (ADAMTS-4) and (indirectly) ADAMTS-5. In addition, a high level of Hif2α decreases the protective role of autophagy, increases the extent of cell death, and activates the RUNX2 and IHH pathways that further contribute to cartilage matrix degradation. The different classes of proteinases that are activated by these cytokines in OA are discussed in more detail later.
Classes of Proteinases (Metalloproteinases, Aggrecanases, Serine and Cysteine Proteases)
In OA, the synthesis and secretion of matrix-degrading enzymes by chondrocytes markedly increases.104,115,140–142 There are four classes of proteases, which are grouped by the catalytic mechanisms of peptide bond cleavage: metalloproteinases, cysteine proteinases, serine proteinases, and aspartyl proteinases. Of these, the first three have clearly defined roles in the degradation of cartilage during the progression of OA.
Metalloproteinases
Metalloproteinases have an enzymatic site that requires a metal ion (often zinc) for activity. Cartilage contains two families of metalloproteinases, namely ADAMTSs and MMPs. Early cartilage degeneration in OA is most likely the result of metalloproteinase enzymatic activity. Both families of metalloproteinases are upregulated in osteoarthritic cartilage, and both are highly expressed in OA cartilage at lesional sites. As the MMPs and ADAMTSs play major roles in the degradation of cartilage extracellular matrix, several MMPs and ADAMTSs are candidate targets for disease modification.143
The control of metalloproteinase activity in OA is complex, with regulation occurring at three different levels: synthesis and secretion, activation of latent enzyme, and inactivation by proteinase inhibitors.144 Most metalloproteinases are not constitutively expressed, but their transcription is induced after stimulation with cytokine and growth factor signaling. Once transcribed, the transcript stability and translation of several metalloproteinases are regulated by microRNA (miR). For example, microRNA-27b (miR-27b) regulates MMP-13 expression.145 Also, miR-140 levels are decreased in OA, which reduces its repression of ADAMTS-5.146 Several of these miRNAs are controlled by the same cytokines and growth factors that maintain cartilage homeostasis including IL-1 and TGF-β. Although the miR field is still in its infancy, it is already evident that several miRs are involved in the pathogenesis of OA by affecting transcript stability and protein translation. Once translated into proteins, almost all MMPs are expressed as inactive proenzymes (zymogens) that require further processing for full proteolytic activity. Most MMPs contain an N-terminal prodomain that blocks or otherwise inhibits the catalytic site. The primary MMP activators are serine- and cysteine-dependent proteases (e.g., the plasminogen/plasmin cascade or furin-like proprotein convertases and cathepsin B, respectively), as well as membrane-type MMPs.118,147,148 Activated MMPs can then be inactivated nonspecifically by α2 macroglobulin and with more specificity by the tissue inhibitor of metalloproteinase (TIMP) family of proteins.
MMPs have historically been further divided into three groups on the basis of their substrate specificities; collagenases cleave across all three chains of the native triple-helical collagens, whereas gelatinases preferably cleave denatured collagen and stromelysins have much broader substrate specificities.149 However, there is considerable overlap in substrates between these classifications; for example, MMP-1 (interstitial collagenase), MMP-3 (stromelysin 1), and MMP-13 (collagenase-3) are all capable of cleaving aggrecan core protein.150 MMPs can degrade other cartilage extracellular matrix molecules in addition to collagen. If combined with plasmin, which has the capability of activating many MMPs, MMPs can rapidly destroy cartilage altogether.
Collagenases
Collagenases typically make the first cleavage in triple-helical collagen, allowing its further degradation by other proteases. Degradation of collagens is thought to be the first irreversible step in the pathogenesis of OA because it significantly reduces the mechanical properties of cartilage. The best-studied MMPs capable of cleaving native collagens are MMP-13 and MMP-1, although MMP-8 and MMP-28 are also involved in breaking down type II collagen. Of the MMPs that degrade native collagen, MMP-13 may be the most important in OA because it preferentially degrades type II collagen.151 Mice deficient in MMP-13 resist developing OA and have reduced cartilage erosion even after proteoglycan loss.152 Expression of MMP-13 greatly increases in OA153; overall collagenase activity also markedly increases in human osteoarthritic cartilage cultures, suggesting that it is a major factor in OA progression and cartilage matrix degradation.154,155 The resultant collagen fragments may then be susceptible to further cleavage by such other enzymes as MMP-2 (gelatinase A), MMP-9 (gelatinase B), MMP-3, and cathepsin B (a cysteine proteinase).
Aggrecanases
The aggrecanases are metalloproteases that belong to a family of extracellular proteases known as ADAMTS.156 Two aggrecanases, ADAMTS-4 and ADAMTS-5, appear to be major enzymes in cartilage degradation in arthritis.157 The roles of these enzymes appear reversed in human and mouse OA—whereas ADAMTS-4 is predominantly associated with aggrecan degradation in human OA, in mouse models of OA ADAMTS-5 is more important.158–160 Recombinant ADAMTS-4 and ADAMTS-5 cleave aggrecan at five distinct sites along the core protein, and all resultant fragments have been identified in cartilage explants undergoing matrix degradation. The G1 region of aggrecan is highly resistant to proteases; however, a glutamate-alanine bond within the extended region between G1 and G2 is remarkably susceptible to proteolytic degradation.161 Aggrecanase proteolytic activity can be modulated by altered expression, by activation by proteolytic cleavage at a furin-sensitive site, by binding to the aggrecan substrate through the C-terminal thrombospondin motif, by activation through post-translational processing of a portion of the C-terminus, and by inhibition of activity by the endogenous inhibitor, tissue inhibitor of metalloproteinase 3 (TIMP-3). ADAMTS-4 and ADAMTS-5 activity is also detected in joint capsule and synovium and may be upregulated in arthritic synovium at either the message level or through posttranslational processing. In addition to ADAMTS-4 and ADAMTS-5, several MMPs are also capable of cleaving aggrecan in vitro (MMP-1, MMP-2, MMP-3, MMP-7, MMP-8, MMP-9, MMP-13, and MMP-28), as are ADAMTS-1, ADAMTS-8, ADAMTS-9, and ADAMTS-15.162 It has been shown that ADAMTS-7 and ADAMTS-12 both bind to and degrade COMP (a prominent noncollagenous protein in cartilage) and that the latter is highly upregulated in osteoarthritic cartilage.163,164 ADAMTS-4, ADAMTS-19, and ADAMTS-20 also have been shown to degrade COMP in vitro; however, their in vivo activity in osteoarthritis has yet to be determined.165,166
A specific hyaluronidase has not been found in articular cartilage matrix, but one or several lysosomal enzymes that can cleave both hyaluronic acid and chondroitin-6-sulfate have been implicated.147 There are six or seven potential hyaluronidases in the human genome, and of these, hyaluronidase-1, hyaluronidase-2, hyaluronidase-3, and PH-20 are likely to be active in cartilage.167 However, experimental evidence to date suggests that these enzymes are either exclusively lysosomal or can exist both in the lysozome and at the plasma membrane. Thus although there is convincing evidence of extracellular degradation of hyaluronan, the precise role of the hyaluronidases in this process has not yet been clearly identified. The decrease in chondroitin sulfate chain length in osteoarthritic cartilage may be due to digestion by synovial fluid hyaluronidase, which may diffuse into the matrix as its permeability increases.103 Evidence to support this theory is the finding that the hyaluronic acid concentration in OA cartilage is low, even though its rate of synthesis is considerably greater than normal.104,115 These degradative enzymes serve to disrupt the proteoglycan aggregate. The early result of the MMP-induced tissue degradation is thinning of the collagen fibers, loosening of the tight collagen network, and the consequent cartilage matrix swelling seen in OA.
Enzyme Inhibitors (Tissue Inhibitor of Metalloproteinases, Plasminogen Activator Inhibitor-1)
The balance of active and latent enzymes is controlled to some extent by at least two enzyme inhibitors: tissue inhibitor of metalloproteinases (TIMP) and plasminogen activator inhibitor-1 (PAI-1).141,168,169 TIMP and PAI-1 are synthesized in increased amounts under the regulation of transforming growth factor-β.170–172 If insufficient concentrations of or degraded TIMP or PAI-1 are present in the matrix along with active enzymes, then increased matrix degradation occurs. Expression profiling of all known members of the MMP, ADAMTS, and tissue inhibitor of metalloproteinases (TIMP) gene families in normal cartilage and cartilage from patients with OA has revealed that several members are regulated in OA. Genes that showed increased expression in OA were MMP-2, MMP-9, MMP-13, MMP-16, MMP-28, ADAMTS-2, ADAMTS-12, ADAMTS-14, ADAMTS-16, and TIMP-3 (all at P < .05). Genes with decreased expression in OA were MMP-1, MMP-3, MMP-10; ADAMTS-1, ADAMTS-5, ADAMTS-9, and ADAMTS-15; and TIMP-1 and TIMP-4.173 These results illustrate the complexity of the events that occur within the extracellular matrix regarding regulation of tissue-degrading enzymes.
Alterations in Matrix Synthesis
Much of what is known about changes in the extracellular matrix in early OA comes from animal models (e.g., the rabbit partial meniscectomy model, the canine anterior cruciate ligament–deficient model).174,175 These animal models represent secondary OA, produced by internal derangement of the joint, and therefore may not precisely simulate the state of affairs in primary OA. In a recent review, models for spontaneous osteoarthritis were reviewed and their preclinical utility was compared.176 Of particular interest is an iodoacetate model, validated as the first pain model of osteoarthritis, in which intra-articular injection of iodoacetate in rats has been shown to lead to cartilage degeneration associated with pain and manifesting as time- and concentration-dependent alterations in hindlimb weight bearing.177,178 That not all animal models of osteoarthritis are equivalent becomes clear when one considers the differences in therapeutic response between young and old animals and between spontaneous and surgical models.179
In the dog model, the first alteration seen within days after joint destabilization is an increase in cartilage water content.180 Initially, water content increases locally, in the tibial plateau and femoral condyle cartilage, but it soon spreads to the entire joint cartilage. Proteoglycans are more readily extractable from the matrix of experimental animals than from that of controls, and this is reflected in the serum biomarker profile after acute injury in patients.181 These matrix changes are also seen in spontaneously occurring dog and steer OA and in experimentally induced rabbit OA.180,182,183 The increase in water content in osteoarthritic cartilage is due to loss of the collagen network’s elastic restraint, enabling the hydrophilic proteoglycans to swell more than normally.184 In early-stage OA, proteoglycan concentration may increase and the cartilage may consequently become thicker than normal and exhibit increased staining for proteoglycans.185–187 Shortly after the increase in cartilage water, newly synthesized proteoglycans are characterized by a higher proportion of chondroitin sulfate and a lower proportion of keratan sulfate and proteoglycan aggregation is impaired.175,180 These abnormal changes in extracellular matrix occur before fibrillation or any other gross morphologic changes are evident and result in a generalized decrease in stiffness that occurs in grossly normal cartilage adjacent to fibrillated areas.188 As OA progresses, focal cartilage ulcerations develop. Proteoglycan loss is accompanied by a decrease in its ability to aggregate, persistence in abnormal glycosaminoglycan composition, and a decrease in chondroitin sulfate chain length. Once proteoglycan loss reaches a critical threshold, water content, which initially increased, falls below normal.189
Chondrocyte Senescence
The term senescence was originally coined to describe the replicative limit of primary cells cultured in vitro, and the concept has evolved to include premature senescence or senescence-like changes in differentiated postmitotic cells. Senescent cells have a unique phenotype that affects both the proteins expressed by senescent cells and their responses to extracellular stimuli. Furthermore, the impact of senescence is not limited to the cells undergoing senescence; rather, the senescent cells have an impact on surrounding cells through the paracrine effects of the altered protein secretions.190
Premature chondrocyte senescence in OA is thought to be enhanced by oxidative injury.191 The inflammatory cytokines IL-1β and TNF that are active in OA induce oxidative stress in chondrocytes, thereby contributing to the potential for oxidative injury that leads to senescence. Oxidative injury is also induced with excessive mechanical shear and injurious loading of explants, and this may contribute to the accumulation of senescent chondrocytes in aged cartilage.192 TGF-β can influence both growth arrest and senescence in cultured cells, and senescent cells can overexpress TGF-β, but this effect has mostly been demonstrated in endothelial cells and not cells of mesenchymal origin. In summary, although it is clearly evident that many of the cellular mechanisms controlling senescence are also active in disease progression including OA, a causal link between senescence and OA has yet to be identified.
Biomechanics and Disease Mechanisms of Osteoarthritis
Biomechanical Changes
Two long-standing biomechanical theories of the pathogenesis of OA hold that mechanical stresses injure chondrocytes, causing them to release degradative enzymes, and that mechanical stresses initially damage the collagen network (as opposed to the cells per se)193,194—in either case, the result is that matrix breaks down. Extracellular matrix breakdown in osteoarthritic cartilage leads to (1) loss of compressive stiffness and elasticity, resulting in greater mechanical stress on chondrocytes; and (2) an increase in hydraulic permeability, resulting in loss of interstitial fluid during compression and increased diffusion of solutes through the matrix (including the movement of degradative enzymes and their inhibitors). One important consequence is disruption of normal fluid film joint lubrication and loading dynamics due to alterations in inflammatory synovial fluid.195–197 Joint friction, wear, lubrication, and contact mechanics are further negatively affected by the loss of cartilage proteoglycans and superficial zone protein (also called lubricin).198–202
Response of Cartilage to Mechanical Injury
The response of normal articular cartilage to injury typically results in suboptimal repair; these injuries can often result in secondary OA.203,204 Articular cartilage produces a repair tissue with neither the original structure nor properties of normal cartilage.205–208 Chondrocytes in areas surrounding an injured zone are unable to migrate, proliferate, repopulate, or regenerate repair tissue with similar structure, function, and biomechanical properties of normal hyaline cartilage.189,205,209
That articular cartilage lacks regenerative power has a long history of documentation.210 In 1851 Redfern reported that articular cartilage wounds healed with fibrous tissue, which he believed arose from chondrocyte intercellular substance.211 Fisher and Ito in the 1920s proposed that cartilage repair is effected by fibrous tissue resulting from proliferation of cells from bone marrow, synovial membrane, and occasionally surrounding articular cartilage.212,213 It was later observed that the fibrous tissue regenerate subsequently transforms into fibrocartilage, with occasional foci of imperfect hyaline cartilage.214–217 The common findings of these investigators were that articular cartilage lacks regenerative potential and that the regenerative fibrous tissue and fibrocartilage tissue must have originated from undifferentiated mesenchymal tissue arising from bone marrow, synovium, or the superficial layer of articular cartilage.210
One reason the reparative process of cartilage significantly differs from those of other tissues is that it is avascular. The healing response in vascularized tissues consists of three main phases: necrosis, inflammation, and repair.203,208 Cartilage undergoes the initial phase of necrosis in response to injury, although typically less cell death occurs than in vascularized tissues because of chondrocytes’ relative insensitivity to hypoxia.203,208 The inflammatory phase, primarily mediated (in other tissues) by the vascular system, is largely absent in partial-thickness injuries (i.e., lesions that do not cross the tidemark), and the repair phase is severely limited given the lack of vascularity and a preceding inflammatory response. Thus no local hyperemia results, no fibrin network is produced, no subsequent clot develops to act as a scaffold for the ingrowth of repair tissue, no mediators nor cytokines are released that can stimulate cellular migration and proliferation, and no inflammatory cells, which have mitotic and reparative potential, are recruited.189,208 In lesions that do not cross the tidemark, the burden of repair falls on the chondrocytes208 in a process that has been termed intrinsic repair.218 Although fetal cartilage is capable of mitotic activity and replication, adult chondrocytes have little potential for replication and intrinsic repair.216,217 Articular cartilage lesions that cross the tidemark may undergo extrinsic repair via differentiation and proliferation of mesenchymal stem cells from para-articular connective tissues; typically, however, a fibrocartilaginous regenerate results.218
There are three categories of articular cartilage injury: (1) microdamage or repetitive trauma to the matrix and cells; (2) partial-thickness or superficial injuries or chondral fractures, articular surface injuries that do not penetrate the subchondral plate; and (3) osteochondral (full-thickness or deep penetrating) injuries, which extend through the tidemark and into the underlying subchondral bone.203,205,208 The host response to each type of injury differs in both timing and quality of repair.
Microdamage to the chondrocytes and/or extracellular matrix without gross disruption of the articular surface can be caused by a single severe impact or repetitive blunt trauma.203,205,208 Repetitive loading of rabbit cartilage produces a surface loss of proteoglycans and an increase in chondrocyte metabolic activity.219 Proteoglycans become more easily extractable from the articular cartilage, with a greater percentage of nonaggregated forms.189 The cellular, metabolic, and biochemical changes after repetitive blunt trauma resemble those in the early stages of OA: increased hydration; cellular degeneration and/or death; disruption of the collagen ultrastructure resulting in marked variation in the size and arrangements of fibers, fissuring and ulceration of the articular surface, thickening of the subchondral bone, and softening of the cartilage with loss of its compressive and tensile stiffness.189,205,220–223 Trauma induces the release of degradative enzymes and proinflammatory factors (e.g., nitric oxide, TNF, IL-1) that frequently cause degradation of the surrounding matrix.219,224,225 Eventually the material properties of the cartilage are altered—cartilage matrix thins and subchondral bone stiffens—which in turn often accelerate the degenerative process.189 The point at which accumulated microdamage becomes irreversible is unknown, although it has been demonstrated that lost proteoglycans and matrix components may be restored if damage to chondrocytes and the collagen network is limited and the repetitive trauma halted.205
Necrosis of neighboring chondrocytes follows chondral fractures and superficial lacerations, injuries that do not cross the tidemark.203,208,226 Within 48 to 72 hours, surviving chondrocytes bordering the defect exhibit increased synthesis rates of extracellular matrix molecules and type II collagen, sometimes accompanied by cell proliferation and formation of clusters or clones in the periphery of the injured zone.189,208,226,227 The increased metabolism and mitotic activity is transient, however, and is followed by a fall in metabolic rate back to normal levels, typically resulting in a suboptimal repair.189,208 Chondrocytes proliferating on the border of the injured zone do not migrate into the defect, which remains unfilled by the newly synthesized matrix.189,205,209 In some cases, superficial lacerations in otherwise normal joints may not progress to full-thickness loss of cartilage or OA.189
Lesions that cross the articular cartilage tidemark and disrupt the underlying subchondral plate elicit the three-phase repair response normally encountered in vascularized tissues. A hematoma forms in the defect that becomes organized into a fibrin clot, activating an inflammatory response. Transformation of the fibrin clot into vascular fibroblastic repair tissue208,227 is accompanied by release of cytokines important in stimulating a repair response (e.g., TGF-β, platelet-derived growth factor, insulin-like growth factor, BMPs).206 These cytokines help set in motion the recruitment, proliferation, and differentiation of undifferentiated cells into a fibrin network that serves as a scaffold for fibrocartilaginous repair tissue.206,228,229 The origin of these mesenchymal stem cells has been determined to be the underlying bone marrow rather than the adjacent residual articular surface.227,229,230 These cells progressively differentiate into chondroblasts, chondrocytes, and osteoblasts and synthesize cartilage and bone matrices. At 6 to 8 weeks postinjury, the repair tissue contains a high proportion of chondrocyte-like cells surrounded by a matrix consisting of proteoglycans and type II collagen, with a lesser amount of type I collagen.230–232 Cells in the deeper layers of the defect differentiate into osteoblasts and subsequently undergo endochondral ossification to heal the subchondral bone defect.189
This regenerative tissue eventually undergoes a transformation to a more fibrocartilaginous repair accompanied by a shift in the synthesis of collagen from type II to type I.209,226,228,229,232 Typically, within 1 year from injury, the repair tissue resembles a mixture of fibrocartilage and hyaline cartilage, with a substantial component (20% to 40%) of type I collagen.231 The size of the osteochondral defect is an important factor in the quality of repair; as a general rule, the smaller the defect, the better the repair.233 Depending on the joint, there exists a critical size defect that will not repair. Fibrocartilaginous repair is susceptible to early degenerative changes because it lacks the biomechanical properties to withstand normal physiologic joint loads.229,231
Mechanotransduction and Gene Expression
Chondrocytes can sense and respond to mechanical stimuli via several regulatory pathways (e.g., upstream signaling, transcription, translation, post-translational modification, vesicular transport).234 Chondrocytes can remodel extracellular matrix in response to alterations in functional demand, as physical forces influence the synthesis, assembly, and degradation of the extracellular cartilage matrix. High magnitude or duration loads can also cause chondrocyte death and collagen damage, and chondrocytes in the superficial zone appear to be more vulnerable to load-induced injury than those in the middle and deep zones.235,236 Normal stimuli help chondrocytes maintain the extracellular matrix; abnormal stimuli can disrupt this balance.
Mechanotransduction influences the cell-mediated feedback between physical stimuli, the molecular structure of newly synthesized matrix molecules, and the resulting biomechanical tissue properties.237 Cell-matrix interactions via integrins are believed to be one of the important mediators in mechanotransduction in chondrocytes. In a study on the expression of COMP to long-term cyclic compression, it was found that uniaxial unconfined dynamic compression significantly upregulated COMP expression, which could be blocked by incubation with anti-α1-integrin antibodies.238 Studies of bovine articular cartilage explants showed that cyclic loading increased protein synthesis by as much as 50% above free-swelling controls and had an inhibitory influence on proteoglycan synthesis, whereas static compression reduced biosynthetic activity.239 Fibronectin and COMP were the most affected noncollagenous extracellular proteins; static compression caused a significant increase in fibronectin synthesis versus free-swelling control levels, and cyclic compression caused a significant increase in synthesis of COMP and fibronectin.
Human articular chondrocytes use the α5β1 integrin as a mechanoreceptor. Mechanical stimulation initiates a signal cascade involving stretch-activated ion channels, the actin cytoskeleton, and focal adhesion complex molecules.240 The result is an anabolic response, manifested by increased aggrecan and decreased MMP-3 expression. Mechanical stimulation also activates Rho and Rho kinase pathways, which are linked to changes in the actin cytoskeleton.241,242 Stimulation of the Rho/ROCK pathway in this context is an anabolic stimulus that leads to nuclear translocation and activation of Sox9, which is a “master regulator” of cartilage gene expression.243 Indian hedgehog (Ihh) protein is a key signaling molecule that controls chondrocyte proliferation and differentiation and may also be an essential mediator of mechanotransduction in cartilage: Cyclic mechanical stress was shown to induce Ihh expression by chondrocytes.244
Dysregulation of these anabolic pathways is thought to contribute to the progression of OA. For example, although the α5β1 integrin is also mechanosensitive in OA chondrocytes, the downstream signaling pathways differ, leading instead to increased cartilage breakdown.245 Integrins and integrin-associated signaling pathways are at least partly regulated by mechanical stimulation by activation of plasma membrane apamin-sensitive Ca2+-activated K+ channels; the result is membrane hyperpolarization after cyclic mechanical stimulation.246 Chondrocytes from normal articular cartilage exhibit membrane hyperpolarization response to cyclic pressure-induced strain, whereas chondrocytes from osteoarthritic cartilage respond by membrane depolarization and exhibit no changes in aggrecan or MMP-3 messenger RNA following mechanical stimulation.247,248 The different signaling pathways responding to mechanical stimulation in healthy versus OA chondrocytes may affect the disease outcome.
In addition to cell and matrix deformation, fluid flow is also sensed by chondrocytes. A study using a tissue shear-loading model to uncouple fluid flow from cell and matrix deformation demonstrated that deformation of cells and pericellular matrix alone stimulated protein and proteoglycan synthesis.249

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


