86 Overlap Syndromes
Apoptotic modification of molecules often renders them more antigenic.
The evolution of symptoms may be associated with molecular mimicry and epitope spreading.
About 50% of UCTDs remain undifferentiated.
The most common presentation of MCTD is Raynaud’s phenomenon.
All six classic AICTDs are descriptive syndromes without a “gold standard” for diagnosis. The diagnosis of a well-differentiated AICTD is usually readily apparent without recourse to extensive investigations. However, in the early stages, there are often common features such as Raynaud’s phenomenon, arthralgias, myalgias, esophageal dysfunction, and positive tests for antinuclear antibodies (ANA). In such cases the diagnosis is not always so obvious; this is often referred to as undifferentiated connective tissue disease (UCTD).1 About 35% of such patients have clinical overlap syndromes, whereas most differentiate into a clinical picture consistent with the traditional description of an AICTD. In some instances one AICTD evolves into another AICTD over time.
The propensity for differentiation into a classic AICTD or the maintenance of an overlap state is often associated with distinctive serologic profiles and major histocompatibility (MHC) linkages. Although most rheumatologists generally feel more comfortable thinking in terms of the classic AICTD paradigms, a case can be advanced for using serologic profiles and human leukocyte antigen (HLA) typing to better understand the clinical features and prognoses. In this respect a careful analysis of the overlap syndromes and their serologic associations has provided insights for understanding the clinical heterogeneity of the AICTDs.2 Researchers have reported numerous clinical correlations of autoantibodies (Table 86-1).
Table 86-1 Correlations of Autoantibodies with Clinical Features
| Autoantigen | Clinical Associations |
|---|---|
| Rheumatoid factor | RA, erosive arthritis, cryoglobulinemia |
| Anticyclic citrullinated protein | RA |
| Nucleosome | SLE, Scl, MCTD |
| Proteasome | SLE, PM/DM, Sjögren’s syndrome, multiple sclerosis |
| Sm snRNP | SLE |
| Histones H1, H2A, H2B, H3, H4 | SLE, UCTD, RA, PBC, generalized morphea |
| Ribosomal P | SLE psychosis |
| dsDNA | SLE, glomerulonephritis, vasculitis |
| ACL/β2-glycoprotein | SLE, thrombosis, thrombocytopenia, miscarriages |
| β2-glycoprotein–independent ACL | MCTD (not associated with APL syndrome) |
| 68-kD peptide of U1-RNP | MCTD, Raynaud’s, pulmonary hypertension |
| U1 snRNP | MCTD, SLE, PM |
| hnRNP-A2 (also called RA-33) | MCTD, RA, erosive arthritis in SLE and Scl |
| Ro/La | Sjögren’s, SLE, congenital heart block, photosensitivity, PBC |
| Fodrin | Sjögren’s, glaucoma, moyamoya disease |
| Platelet-derived growth factor | Diffuse and limited Scl |
| Topoisomerase I (Scl-70) | Diffuse Scl with prominent organ involvement |
| Centromere | Limited Scl, CREST, Raynaud’s, pulmonary hypertension, PBC |
| Th/To | Limited Scl |
| U3-snRNP | Limited Scl |
| hnRNP-I | Scl (early diffuse and limited) |
| RNA polymerases I and III | Scl (diffuse with renovascular hypertension) |
| Fibrillarin | Severe generalized Scl |
| Ku | Myositis overlap, primary pulmonary hypertension, Graves’ disease |
| U5-snRNP | Myositis overlap |
| PM/Scl | Myositis overlap with arthritis, skin lesions, mechanic’s hands |
| Signal recognition particle | Myositis overlap (severe course with cardiac disease) |
| Antisynthetases (Jo-1, PL-7, PL-12) | Myositis overlap with arthritis and interstitial lung disease |
| Mi-2 | Dermatomyositis |
| Proteinase-3 | Granulomatosis with polyangiitis (formerly Wegener’s granulomatosis), pulmonary capillaritis |
| Myeloperoxidase | Churg-Straus, pauci-immune glomerulonephritis |
| Endothelial cell | Pulmonary hypertension, severe digital gangrene |
| α-Enolase | Behçet’s, RA, MCTD, Scl, Takayasu’s |
| Angiotensin-converting enzyme 2 | AICTDs with vasculopathy |
ACL, anticardiolipin; AICTDs, autoimmune connective tissue diseases; APL, antiphospholipid syndrome; CREST, syndrome of calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia; DM, dermatomyositis; hn, heterogeneous nuclear; MCTD, mixed connective tissue disease; PBC, primary biliary cirrhosis; PM, polymyositis; RA, rheumatoid arthritis; RNP, ribonucleoprotein particle; Scl, scleroderma; SLE, systemic lupus erythematosus; sn, small nuclear; UCTD, undifferentiated connective tissue disease.
Epidemiology
The reported prevalence of AICTDs is variable, depending on methodology, nature of referral bias, and ethnicity.3 It is generally accepted that Sjögren’s syndrome has the highest prevalence (0.5 to 3.6%), with SLE being much lower at about 15 to 50 per 100,000. Scleroderma, polymyositis, and dermatomyositis are relatively rare AICTDs, occurring in fewer than 10 per 100,000. Experts are increasingly realizing that overlap syndromes of scleroderma and myositis are more common than the “pure” forms of the disease.4 There are no epidemiology studies of overlap syndromes, apart from Japan, where the reported prevalence of mixed connective tissue disease (MCTD) was 2.7 per 100,000. The syndrome of MCTD usually occurs as an isolated finding, but there are reports of a familial occurrence. Unlike SLE, precipitation by sun exposure has not been described in patients with MCTD. Likewise, drug exposure has not been related to the onset of MCTD, although a transient appearance of anti-RNP antibodies has been seen at the initiation of procainamide therapy.5 Vinyl chloride and silica are the only environmental agents that have been associated with MCTD so far.
Autoimmunity in Overlap Syndromes
Compelling evidence indicates that autoimmunity is often antigen driven by components of subcellular particles, in particular spliceosomes, nucleosomes, and proteasomes.6
Autoimmunity to Spliceosomal Components
Certain components of the spliceosome are common targets of autoimmunity in the AICTDs.7 Furthermore, it appears that post-translational modifications of these molecules, as occurs during apoptosis, are often associated with increased immunogenicity.8 Spliceosomes are complex nuclear particles made up of some 300 distinct proteins and 5 RNAs, which are involved in the processing of premessenger RNA (pre-mRNA) into mature “spliced RNA.”9 Two major spliceosomal subunits are antigenic targets in autoimmunity: (1) small nuclear ribonucleoprotein protein particles (snRNPs) and (2) heterogeneous nuclear RNP particles (hnRNPs).
The snRNPs contain small RNA species ranging in size from 80 to 350 nucleotides that are complexed with proteins.6 These RNAs contain a high content of uridine and are therefore called U-RNAs; 5 different U-RNAs were defined on the basis of immunoprecipitation (U1, U2, U4, U5, and U6). Autoantibodies to these complexes are mainly directed to the protein components. Anti-Sm antibodies precipitate five proteins with molecular weights of 28,000 (B′B), 16,000 (D), 13,000 (E), 12,000 (F), and 11,000 (G); five of these polypeptides are common to the U1, U2, U4, U5, and U6 RNAs. Anti-RNP antibodies precipitate three proteins with molecular weights of 68,000 (70K), 33,000 (A′), and 22,000 (C); these polypeptides are uniquely associated with U1 RNA (Figure 86-1).The clinical correlates considered to be distinctive of MCTD are associated with the 70-kD specificity with an immunodominant epitope embracing amino acid residue 125 flanked by important conformational residues at positions 119-126 (see Figure 86-1). On the other hand, SLE is associated with anti-Sm antibodies.
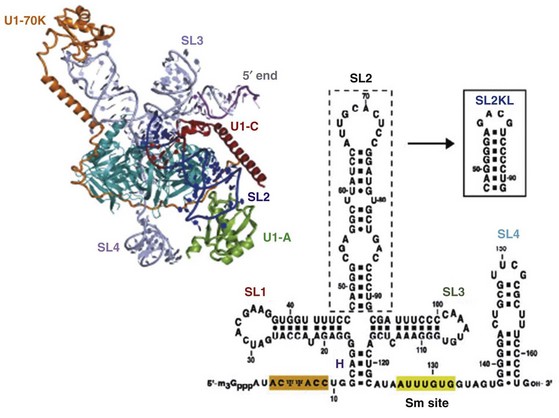
Figure 86-1 The spliceosome is made up of five small nuclear RNAs (snRNAs) complexed with proteins to form a small nuclear ribonucleoprotein particle (snRNP). This subcellular structure is responsible for splicing introns from pre-mRNA to form mRNA via a 59-splice recognition site. Antibodies to various spliceosomal constituents are a common feature of autoimmune rheumatic disorders with a tendency to associate with different clinical profiles (see Table 86-1). The U1 small nuclear RNP (U1-snRNP) particle of the spliceosome is composed of U1-RNA, RNP proteins (70 kD, A, and C), and common Sm proteins (B′B, D, E, F, and G). The structure of U1-RNA consists of single-stranded RNA and double-stranded RNA called stem loops (SL) 1, 2, 3, and 4. An electron density map of the functional core of U1-snRNP at 5.5 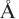 resolution has enabled the spatial visualization of the RNA and placement of the seven Sm proteins, U1-C, and U1-70K into the map. A striking feature is the amino (N)-terminal polypeptide of U1-70K, which extends over a distance of 180
resolution has enabled the spatial visualization of the RNA and placement of the seven Sm proteins, U1-C, and U1-70K into the map. A striking feature is the amino (N)-terminal polypeptide of U1-70K, which extends over a distance of 180 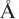 from its RNA binding domain, wraps around the core domain consisting of the seven Sm proteins, and finally contacts U1-C, which is crucial for 59-splice-site recognition.
from its RNA binding domain, wraps around the core domain consisting of the seven Sm proteins, and finally contacts U1-C, which is crucial for 59-splice-site recognition.
(Modified from Newman J: Structural studies of the spliceosome, Curr Opin Struct Biol 20:82–89, 2010; and Pomeranz AD: Crystal structure of human spliceosomal U1 snRNP at 5.5 A resolution, Nature 458:457–480, 2009.)
The hnRNPs are among the most abundant proteins in the eukaryotic cell nucleus. They contain pre-mRNA associated with 30 small proteins that are all structurally related and have molecular weights of 33 to 43 kD. Nine hnRNP core proteins have been designated A1, A2, B1a, B1b, B1c, B2, C1, C2, and C3. An antibody termed anti-RA33, which targets the 33-kD hnRNP-A2, is particularly interesting because it is found in about one-third of sera from patient with RA, SLE, and MCTD.10 It also has associations with patient subsets of erosive arthritis in SLE, scleroderma, and MCTD and predicts the eventual development of RA in patients with early polyarthritis.11 Importantly, this association with anti-RA33 is not seen in scleroderma (sine erosions), PM, or overlaps of PM/Scl or PM/DM. The antigenic epitopes of hnRNP-A2 contain two RNA binding regions at the N-terminal end and a glycine-rich C-terminal region. Certain disease subsets target these two RNA binding regions differently. For instance, RA and SLE sera preferentially react with the complete second RNA binding domain, whereas MCTD sera target an epitope that spans both RNA binding domains.
Autoimmunity to Nucleosomal Components
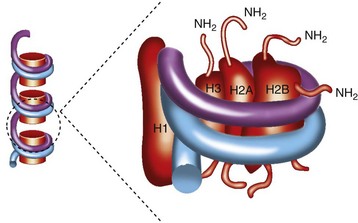
Nucleosomes are the compact building blocks of chromatin and consist of an octamer of two copies of histones H2A, H2B, H3, and H4, wrapped around approximately 146 base pairs of DNA (Figure 86-2). During apoptosis endonucleases cleave chromatin with the liberation of nucleosomal particles. Following the release into the cytoplasm, nucleosomes migrate to the surface of the dying cell12 and thus become accessible to B cell receptors. The development of autoimmunity has been linked to defective phagocytosis of apoptotically released constituents.12 Nucleosomal antibodies are directed to antigenic determinants on the intact nucleosome rather than its individual components, DNA and histones. In a study of 496 patients with 13 different AICTDs and 100 patients with hepatitis C, antinucleosome antibodies were found in the sera of patients with SLE (71.7%), Scl (45.9%), and MCTD (45.0%).13
Autoimmunity to Proteasomal Components
The 26S proteasome is a large subcellular particle involved in the degradation of proteins that have been tagged with ubiquitin, resulting in the generation of peptides for presentation by the MHC class I molecules (Figure 86-3). There is good evidence that it is the target of an autoimmune response in AICTD. Antibodies to proteasomal subunits have been reported in patients with autoimmune myositis, systemic lupus erythematosus, and primary Sjögren’s syndrome. Circulating 20S proteasomes (c20S) subunits appear to have an association with disease activity in MCTD and SLE.14
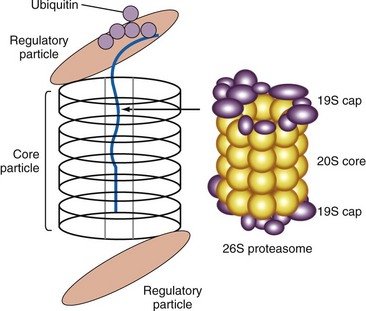
Generation of Autoimmunity
The antibody response to just one component of an intracellular structure such as a spliceosome will result in the uptake of the entire particle by antigen-presenting cells (APCs). Thus all the proteins making up the particle will be subject to antigen processing with potential peptide presentation linked to their affinity to class II HLA antigens. Depending on the polymorphisms of the individual HLA molecules, there will be a diversification of the antibody response to include some of these other antigens. This process is called epitope spreading and is considered pivotal in the development of the linked antibody responses that are observed in different connective tissue diseases (CTDs).15 For instance, it has been shown that the induction of an immune response to one component of a U-RNP complex can induce a diversified autoantibody response to other components of the complex.16 In this way an immune response becomes modified over time, and this change has been associated with changes in the clinical picture.
The interaction between T cell receptors and peptides presented by HLA molecules is a critical event in the generation of autoimmunity. The 70-kD and anti-U1-RNP antibody responses are associated with the HLA DR4 and DR2 phenotype.17 In a transgenic murine model of MCTD, the majority of T cells targeted a limited number of epitopes residing within the RNA binding domain of the 70-kD antigen.18 DNA sequencing of HLA-DB genes has revealed that DR2- and DR4-positive patients share a common set of amino acids in the beta chain at positions 26, 28, 30, 31, 32, 70, and 73 that form a pocket for antigen binding. It is hypothesized that these two HLA subtypes represent a critical genetic specificity for the presentation of antigenic peptides to their cognate T cell receptors. The shared epitope on HLA-DR4/DR2 that is associated with an anti-U1-RNP response is different from the shared epitope associated with HLA-DR4/DR1 in RA patients.19 The 70-kD polypeptide has several different epitopes, the most consistent sequence being KDK DRD RKR RSS RSR.20 This region is preferentially targeted by MCTD sera but not by SLE sera.21 The autoimmune response to the spliceosome in these three disorders is characterized by differential degrees of epitope spreading. The widest range of antibodies, to both snRNP and hnRNP, is seen in SLE; a more restricted antispliceosomal antibody repertoire to snRNP and hnRNP is seen in MCTD; and in RA the antispliceosomal antibody repertoire is restricted to hnRNP.22 In general the autoimmune rheumatic diseases are characterized by the production of autoantibodies that recognize evolutionarily conserved molecules. The mechanism whereby these “hidden” intracellular molecules become autoantigens is an area of ongoing research. The two main theories are apoptotic modification23 and molecular mimicry.24
Proteins modified during apoptosis can be presented to the immune system in ways that bypass tolerance to self-proteins.6 Although rheumatic disease autoantigens are not unified by common structure or function, they have the common feature of becoming clustered and concentrated in the surface blebs of apoptotic cells. A population of smaller blebs contains fragmented endoplasmic reticulum and ribosomes, as well as the ribonucleoprotein Ro. Larger blebs (apoptotic bodies) contain nucleosomal DNA, Ro, La, and the small nuclear ribonucleoproteins.25 During the process of apoptosis several enzyme systems are upregulated with resulting post-translational modifications of the cleaved proteins. These modifications, which include citrullination, phosphorylation, dephosphorylation, transglutamination, and conjugation to ubiquitin, render the molecules more antigenic. For instance, the U1-70K protein is cleaved by the enzyme caspase-3, converting it into a C-terminally truncated fragment, which contains a major B cell epitope that is preferentially recognized by autoimmune sera.26
The initial stimulus for a first antibody response may be a non–self-protein possessing a peptide region that mimics a self-epitope—so-called molecular mimicry. Environmental stressors such as infection, toxins, drugs, and ultraviolet light may, under some circumstances, induce accelerated apoptosis. A critical limitation to molecular mimicry is the necessity for the antigenic sequence to undergo TCR recognition. Helper T lymphocytes (CD4+) usually recognize peptides of 12 to 16 amino acids in the context of HLA class II molecules. However, in some instances smaller peptides may be recognized. They can be more immunostimulatory than the parent ligand. Thus antigen recognition by T cells is highly degenerate and expands the potential for molecular mimicry. The universe of molecules containing a pentapeptide, for example, is much greater than for 12 amino residue peptide. Once an immune response to one component of an immunogenic molecular complex has been elicited, other proteins/epitopes of the complex may become antigenic by the same process of epitope spreading.15
Undifferentiated Connective Tissue Disease
Key Points
Nail-fold capillary microscopy is useful in evaluating the potential pathology.
Antibody profiles are useful in predicting the eventual clinical features:
U1-RNP antibodies predict the differentiation into MCTD.
DNA antibodies predict the differentiation into SLE.
Nucleolar antibodies predict the differentiation into systemic sclerosis (SSc).
Synthetase and PM/Scl antibodies predict the differentiation into a myositis overlap syndrome.
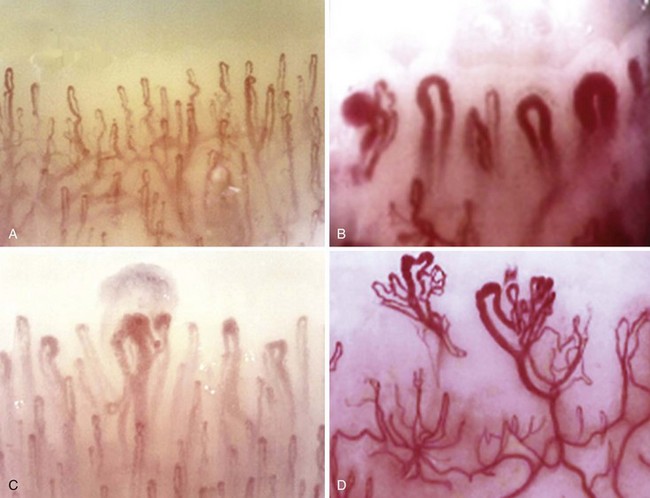
The answer to this question is not always straightforward because fibromyalgia is not a diagnosis of exclusion27; it is a common comorbidity with the well-defined CTDs and is often associated with cold-induced vasospasm.28 An algorithm for diagnosing UCTDs is given in Figure 86-4. In the early stages of a CTD, there may be just one or two suspicious clinical and laboratory features, but a definitive diagnosis cannot always be made. In such cases a working diagnosis of undifferentiated connective tissue disease (UCTD) may be appropriate.29 Most patients with this UCTD have Raynaud’s phenomenon with or without an unexplained polyarthralgia and a positive ANA with usually just a single autoantibody specificity, often anti-Ro and anti-RNP.30 A 5-year follow-up study of 665 patients with UCTD reported that only 34% developed a well-defined CTD (RA—13.1%, Sjögren’s—6.8%, SLE—4.2%, MCTD—4%, Scl—2.8%, systemic vasculitis—3.3%, and PM/DM—0.5%).31 Certain combinations of features are predictive for the development of an established CTD: Polyarthritis plus U1-RNP antibodies predict MCTD, sicca symptoms plus anti-SSA/SSB antibodies predict Sjögren’s syndrome, Raynaud’s phenomenon plus a nucleolar ANA pattern predict Scl, polyarthritis plus high levels of rheumatoid factor (RF) predict RA, and fever/serositis plus a homogeneous ANA pattern or anti-dsDNA antibodies predict progression into SLE (Table 86-2). The identification of a pathologic nail-fold capillary pattern can provide some early indication that the UCTD may progress to systemic sclerosis (SSc) or MCTD (Figure 86-5).32 Low levels of vitamin D are also reported to be a risk factor for the development of UCTDs and should be evaluated and corrected in all such patients.33
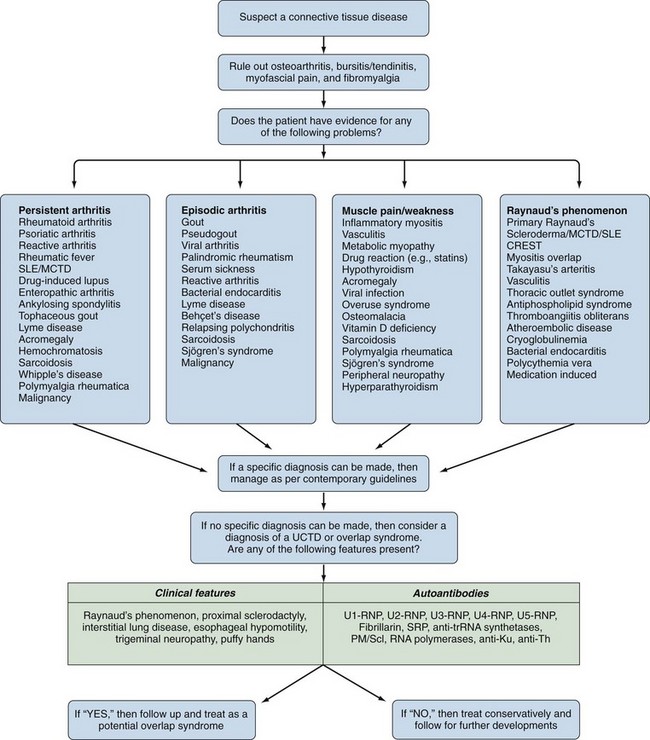
Table 86-2 Disorders Associated with Increased Fibrosis
| Localized |
| Systemic |
Scleroderma Overlaps
Key Points
CREST has a common overlap with primary biliary cirrhosis.
Pulmonary fibrosis and pulmonary hypertension are the main causes of morbidity/mortality.
Several fibrotic conditions may mimic scleroderma (see Table 86-2). Scleroderma itself has a widespread heterogeneity of disease expression ranging from a diffuse cutaneous disease, with a poor prognosis, to a limited cutaneous involvement, with generally a good prognosis. Furthermore, some patients with Scl have a prominent overlap with other connective tissue diseases.34 In many cases, these overlaps occur in patients who do not have prominent skin involvement (sine scleroderma) or with the limited form of the disease—CREST. Approximately 90% of patients with Scl have a positive ANA. Scleroderma-related antibodies include topoisomerase I (Scl-70), anticentromere (ACA), hnPNP-I, RA33, p23, p25, RNA polymerase I (RNAP-1), RNA polymerase III, U1-RNP, PM/Scl, fibrillarin, histone, Ku, endothelial cell, and Th/To35 (see Table 86-1).
A German registry for scleroderma has reported on patterns of organ involvement in two subsets of 1483 SSc patients.36 Limited distal skin involvement (distal to the knee and elbows) was seen in 46% (the lcSSc group), and 33% had progressive widespread scleroderma (rapid involvement of trunk, face, and extremities—the dcSSc group). An overlap syndrome was seen in 11%, and 8% were undifferentiated. The extent of organ involvement varied between subgroups. For instance, musculoskeletal involvement was seen in 68%of the overlap group compared with 57% of the dcSSc group. Pulmonary fibrosis (56%) and pulmonary hypertension (19%) were most common in the dcSSc group, but pulmonary hypertension was seen in 15% of the dcSSc group.
Specific antibody profiles tend to be associated with distinctive patterns of morbidity and mortality.37 Patients possessing anticentromere, anti-U3 snRNP, and anti-Th/To antibodies tend to have the limited form of Scl, whereas anti-Scl-70, ACA, and anti-RNAP are associated with diffuse skin involvement and systemic disease. Anti-PM/Scl antibodies are associated with a myositis/Scl overlap and a tendency to develop pulmonary interstitial disease.38 About 60% of patients with scleroderma have obvious synovitis, and 35% are positive for RF. Erosive arthritis in Scl has an association with anti-RA33; the Scl component in such overlap patients is often an incomplete form of CREST.39 The limited form of scleroderma has a well-documented overlap with primary biliary cirrhosis (PBC). The distinctive antibody association of scleroderma with PBC is antimitochondrial antibodies.40 Conversely, anticentromere antibodies have been found in 10% to 29% of patients with PBC; approximately half developed some features of the CREST syndrome. Hence a serologic overlap between the two syndromes is more prevalent in the clinical overlap. Low-grade muscle involvement is not uncommon in scleroderma, being described in between 50% and 80% of patients. A European review of 114 scleroderma overlap patients reported a 95% PM/Scl antibody positivity41 with 80% having an inflammatory myositis. This “scleromyositis” differed from MCTD by coexistent features of dermatomyositis (myalgia, myositis, Gottron sign, heliotrope rash, calcinosis), but no overlap SLE features, as is characteristic of classic MCTD. Many of these patients had a deforming arthritis of the hands. In general they had a chronic benign course, and most were steroid responsive. Scleroderma lupus overlaps are less common. However, Scl patients often have antinuclear antibodies other than ACA and Scl-70.
Nonscleroderma fibrotic disorder may be mistaken for a scleroderma overlap at initial presentation (see Table 86-2); although these disorders may have some systemic involvement, they seldom exhibit overlap features with other AICTDs.
Nephrogenic systemic fibrosis (NSF) is a fibrotic disorder that develops in some patients following exposure to gadolinium-containing contrast agents; most patients have pre-existing renal disease.42 Histologically there is fibroblast proliferation, thickened collagen bundles, and deposits of mucin, similar to those observed in scleromyxedema. The clinical presentation is a rapid progression with confluence of initially focal areas of indurated skin (Figure 86-6). The face is usually spared, but joint contractures may occur at the elbows and knees, and systemic involvement with pulmonary and neurologic symptoms can develop in refractory cases. NSF is usually nonresponsive to corticosteroids and immunosuppressive therapy.
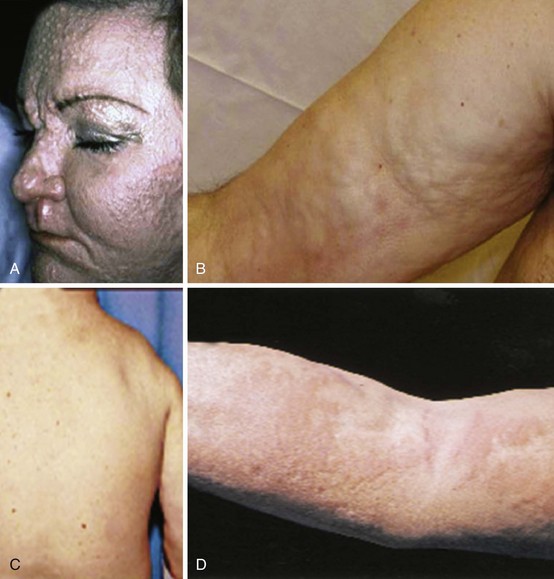
Eosinophilic fasciitis presents with limited scleroderma-like skin changes involving the extremities (see Figure 86-6). The correct diagnosis is suggested by finding a peripheral eosinophilia and a hypergammaglobulinemia.43 The definitive diagnosis is established by a full-thickness skin biopsy that shows a diffuse inflammation of the fascia. Initial treatment is with corticosteroids (prednisone 0.5 to 1 mg/kg) tapering according to the clinical response; some patients need to continue moderate-dose corticosteroids for up to 2 years. Methotrexate and mycophenalate may be used in refractory cases.
Scleromyxedema is characterized by cutaneous mucinosis and is often associated with a gammopathy, usually IgM and light chains.44 The mucinous skin lesions appear as waxy papules on the face, neck, and limbs. If the papules coalesce, it may be mistaken for scleroderma (see Figure 86-6). Systemic involvement may occur with dysphagia, proximal muscle weakness, pulmonary, cardiac, and renal complications. It is difficult to manage; corticosteroids are usually tried initially, in refractory cases some benefit has been reported for intravenous immunoglobulin and thalidomide.
Scleredema is a cutaneous mucinosis that often starts with a febrile episode and resolves spontaneously.45 More chronic scleredema has been associated with paraproteinemias including multiple myeloma and diabetes mellitus. The dermis is thickened with increased collagen glycosylation, as in diabetic stiff skin syndrome. The face and neck are commonly involved, and there is relative sparing of the hands and feet (see Figure 86-6). Systemic organ involvement is rare, but a monoclonal gammopathy is sometimes seen. Such cases need to be worked up for lymphoma. Refractory cases have been helped by local radiotherapy.




