85 Inflammatory Diseases of Muscle and Other Myopathies
History of Inflammatory Muscle Diseases
Inflammatory muscle diseases are a heterogeneous group of systemic autoimmune rheumatic disorders characterized by chronic muscle weakness, muscle fatigue, and mononuclear cell infiltration into skeletal muscle. These disorders were described in the literature more than a century ago as generalized muscle disorders affecting principally the trunk and proximal limb muscles, with or without skin involvement.1–5 It was also recognized that these diseases can range from acute and even fatal to slow, progressive, chronic, insidious conditions, with patterns of relapse and remission. Steiner’s6 summary of myositis cases in 1903 made a clear distinction between idiopathic polymyositis (PM) and other forms of myositis caused by bacteria and parasites,6 and Stertz7 in 1916 first reported an association between dermatomyositis (DM) and internal malignancy.7 At about the same time, Batten8 described the first case of DM with classic histologic features in a child.
Since the 1940s, it has been recognized that PM may occur in the absence of cutaneous lesions, muscle pain, or constitutional symptoms. It may present in an acute, subacute, or chronic insidious form, with some fraction of cases showing systemic features or involvement of organs and tissues.9 The differential diagnosis has been described independently by several investigators, and the most chronic form was differentiated from an adult variety of muscular dystrophy.10–12 Banker and Victor13 noted that DM in children was different and involved a greater degree of vascular inflammation and thrombosis (systemic angiopathy). The first, and still widely used, classification scheme and set of diagnostic criteria for myositis were proposed by Bohan and Peter in 1975.14,15 They include PM and DM but not the later-described subset known as inclusion body myositis (IBM). IBM was later defined by the presence of distinct histopathologic changes including vacuoles and nuclear and cytoplasmic inclusions, as well as by distinct clinical features including resistance to glucocorticoids.16,17 Debate continues whether IBM should be considered an idiopathic inflammatory myopathy (IIM). We have chosen to include information on IBM in this chapter because it is clinically relevant to the differential diagnosis of PM.
Epidemiology
The actual annual incidence of inflammatory myopathy is currently unknown. Because these diseases are so rare, no large-scale epidemiologic studies have been reported; however, several retrospective studies have reported an annual incidence of less than 10 per million individuals18–22 (Table 85-1). This may be an overestimate, given that the Peter and Bohan diagnostic criteria used in these studies did not distinguish IBM as a separate disease entity. The prevalence of IBM has been estimated to be 10.7 per million in the United States, 9.3 per million in Australia, and 4.9 per million in the Netherlands.23–25 The age-adjusted prevalence of IBM for those older than 50 years was reported as 16 to 35 per million.24,25 In some geographic areas, IBM appears to be the most common acquired progressive myopathy, representing 16% to 28% of all inflammatory myopathies.25 There may be referral biases in these studies. The incidence-prevalence studies need to be interpreted cautiously, given that most have not reported confidence intervals for their rates.
The incidence of the various myopathies varies according to ethnicity, age, and gender. Some studies have reported that the incidence of PM is higher in black patients than in white patients.18 IIMs can occur in any age group, from early childhood to late in adult life. The onset of PM is usually in the late teens or older, with the mean age at onset being 50 to 60 years; DM shows two peaks—5 to 15 years and 45 to 65 years. IBM is commonly seen in individuals older than 50 years and is rare in younger adults. Some studies have reported gender-specific incidence rates. For example, in the case of PM and DM, females are more commonly affected than males (ratio > 2 : 1), whereas in IBM, the converse is true (again, >2 : 1 ratio).
Inflammatory myopathies can occur in association with other autoimmune connective tissue diseases such as scleroderma, systemic lupus erythematosus (SLE), rheumatoid arthritis, Sjögren’s syndrome, polyarteritis nodosa, and sarcoidosis. Significant proportions of all myositis patients (11% to 40%) have an associated connective tissue disease.23,26,27 Several studies have also confirmed an association between malignancies and inflammatory myopathies. A 2-decade-long retrospective study of myositis patients revealed about 12% (37/309) of the patients are associated with malignant diseases. A majority (81%) (30/37) of these patients had DM, and the remainder (19%) had PM. In about 68% of these cases malignancy and myositis appeared within 1 year. The most frequent malignancies associated with myositis were breast tumors and adenocarcinomas, and successful treatment of the underlying malignant disease improved the clinical disease course of myositis. Overall survival rate was considerably worse for DM compared with other forms of myositis.28 Similar observations were made in a nationwide cohort study in Taiwan that indicated although the general risk of cancer was increased in IIM patients, cancers of the nasopharynx, lung, and breast tissue were the most likely to be diagnosed in both PM and DM patients. Detection of malignancies most frequently occurred within 1 year of being diagnosed with an IIM, with the likelihood of developing a malignancy decreasing over time.29 Overall, the frequency of malignancies varies widely (4% to 42%) in different studies,19,30 but in general the incidence of malignancy is higher in DM patients than in IBM or PM patients.31 It is difficult to determine the relative risks for a particular malignancy because a variety of malignancies are associated with myositis, and only small numbers of individual malignancies have been reported in any one study.
Etiology of Myositis
Genetic Risk Factors
An association with immune response genes and occasional reports of familial clustering of myositis support the role of genetic factors in these diseases.32–37 Polymorphisms in human leukocyte antigen (HLA) class I and II genes are known genetic risk factors for several autoimmune diseases including myositis, but the mechanisms for these associations remain unclear. One possibility is that because the gene products influence T cell repertoire development, tolerance, and immune responses to foreign agents, certain polymorphisms may be selected on the basis of environmental triggers. It appears that haplotypes HLA-DRB1*0301 and HLA-DQA1*0501 are the strongest known genetic risk factors for all forms of myositis in whites; however, different phenotypes have additional HLA risk and protective factors.37,38 It has been shown that in African-American patients neither DRB1*0301 nor DQA1*0501 is strongly associated with myositis. Instead, the HLA-DRB1*08 allele shows the highest general risk for developing myositis, whereas the HLA-DRB1*14 allele is strongly protective in African-American patients.39 The HLA-B8/DR3/DR52/DQ2 haplotype is found in a significant proportion of IBM patients.40 The risk and protection conferred by HLA associations differ significantly among different ethnic and serologic groups. For example, in some populations (e.g., Koreans, Mesoamericans), there is no association with HLA genes.35 Further, HLA-DRB1*0301, which is a risk factor in whites, is a protective factor in the Japanese population.41 The HLA-DRB1*0301, HLA-DQA1*0501, and HLA-DQB1*0201 alleles are strongly associated with myositis-specific antibodies in PM patients.42 Mechanistic data supporting the role of HLA molecules in disease pathogenesis are, unfortunately, lacking at present. Some studies have reported that maternally derived chimeric cells are present in the peripheral blood and muscle tissues of juvenile DM patients, suggesting that HLA alleles control the occurrence of chimerism and explain the HLA association found in these disorders.43,44 Like other autoimmune disease conditions, myositis is a complex multigenic disorder involving other non-HLA immune response genes (e.g., cytokines and receptors including tumor necrosis factor [TNF], interleukin [IL]-1, and tumor necrosis factor receptor [TNFR]-1); complement components (e.g., C4, C2); immunoglobulin heavy-chain allotypes, and T cell receptors.45 The exact contribution of the genetic component in these disorders is currently unknown, in part because of their rarity, the small number of subjects in any single cohort, and the heterogeneity in disease phenotype. International collaborative efforts are currently under way to address these issues and to identify potential genetic and environmental risk factors in myositis.
Environmental Risk Factors
The temporal association of myositis onset and environmental agents in certain individuals suggests that specific exposures in the context of certain genetic backgrounds can initiate muscle inflammation. Common environmental agents implicated in myositis include infectious organisms such as viruses and bacteria and noninfectious agents such as drugs and food supplements (Table 85-2). For example, enteroviruses (influenza, coxsackievirus, echoviruses) and retroviruses (human T-lymphotropic virus-I) are known to induce muscle inflammation. The myositis associated with enteroviruses usually occurs in children and is generally self-limited. A viral cause is strengthened by the presence of high-titer antiviral antibodies and viral particles in patients’ serum and tissue samples,46,47 as well as the induction of muscle inflammation by enteroviruses in animal models. Attempts to identify virus in the tissues of IIM patients by sensitive techniques such as polymerase chain reaction have failed, leading to doubts about the viral cause of these diseases48 and ruling out continual viral infection as a cause of the ongoing muscle inflammation in these patients. However, it is possible that viruses initially trigger the disease process before being eliminated by the host’s immune response, thus explaining the absence of viral genomes in the myositis muscle tissue. Similarly, some microorganisms such as staphylococci, clostridia, and mycobacteria are known to affect skeletal muscle and cause acute muscle inflammation, but there is no evidence that these organisms actually cause chronic, self-sustaining muscle inflammation.
Table 85-2 Possible Environmental Risk Factors
| Infectious Agents |
| Noninfectious Agents |
BCG, bacille Calmette-Guérin; HIV, human immunodeficiency virus; HTLV-I, human T-lymphotropic virus I.
Parasites such as Toxoplasma gondii, Trypanosoma cruzi, and Borrelia burgdorferi have been implicated in the triggering of IIMs. The evidence in support of a parasitic cause includes the recovery of parasites from some myositis patients and their serologic response to the parasites; improvement in myositis symptoms after treatment with antiparasitic drugs; a histologic picture of inflammation including infiltration of macrophages and CD4 T cells; and induction of myositis after parasitic infection in animal models.49–55 Despite these observations, it is difficult to establish a direct link between any parasitic infections and myositis in human patients because there is often no history of antecedent parasitic infection.
Ultraviolet (UV) light irradiation is likely to be a risk factor for the development of DM because epidemiologic data have demonstrated a latitude gradient of PM and DM, with the latter being more frequent closer to the equator and the former being more frequent in northern countries. The ratio between PM and DM is associated with a latitude gradient and is directly correlated with UV light irradiation. This observed correlation is particularly strong in a subset of DM patients with anti–Mi-2 autoantibodies, indicating that UV light may be an environmental risk factor for its development. The association between UV light exposure and subtype of myositis suggests that UV light is an exogenous modifier that can influence the clinical phenotype in PM and DM.56
It appears that malignancy is an additional risk factor for the development of myositis, and there is a strong association between DM and malignancies. This early clinical observation has been confirmed in epidemiologic studies.30,57 With regard to PM and IBM, the association with malignancy is less convincing. The increased risk of malignancy associated with DM has been established both at the time of DM diagnosis and more than 10 years after diagnosis. The pathophysiologic mechanism for the association between malignancy and DM has not been clarified, but there could be several explanations. The strong association between malignancy and the onset of DM indicates that the latter could be a paramalignant phenomenon; that is, the development of myositis is a consequence of the malignancy (related to autoantigens), or the malignancy and DM share disease mechanisms. Thus the molecular mechanisms underlying this unique association are currently unclear. However, there is some evidence that removal of a tumor sometimes results in amelioration of muscle weakness, and tumor reappearance sometimes coincides with muscle weakness, suggesting that these two are linked.28 A recent report has shed some light on this connection by showing that myositis-specific antigens are highly expressed in cancer tissues, as well as in regenerating muscle cells of myositis patients.58,59 The authors propose that in cancer-associated myositis, an autoimmune response directed against cancer cross-reacts with regenerating muscle cells, enabling a feed-forward loop of tissue damage and antigen selection.60 This association must be explored further because cancer-associated myositis patients almost never develop myositis-specific autoantibodies, which are protective for the development of cancer. For malignancies that develop during established disease, the potential explanations include the presence of chronic inflammation or prolonged immunosuppressive treatment, which could contribute to the development of malignancy.
A recent report noted a novel association of myositis with hypertension, diabetes, and ischemic heart disease. The prevalence of hypertension and diabetes in this population was 62% and 29%, respectively, considerably higher than the background prevalence of 9.4% and 4%. These authors suggest that hypertension and ischemic heart disease were more likely to be present before the diagnosis of myositis, whereas hypertension and diabetes occurred more frequently following the diagnosis of myositis in DM patients in comparison with PM or IBM patients, suggesting that it is essential to perform a comprehensive assessment of vascular risk factors in these patients.61 The same group also reported that patients with IIM are at 75% increased risk for mortality, and cardiovascular diseases followed by infection and malignancy account for the commonest causes of death.62
Mimics of Myositis
A variety of insults induce the clinical and pathologic spectrum that mimics myositis in some individuals (see Table 85-2). A number of drugs are known to cause a myopathy that closely mimics myositis. For example, d-penicillamine causes clinically and histologically indistinguishable IIM.63 Likewise, commonly used lipid-lowering drugs such as statins (e.g., atorvastatin, lovastatin) can cause a myopathy that resembles inflammatory myositis. These agents inhibit 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, a rate-limiting enzyme involved in the conversion of HMG-CoA to mevalonic acid, thereby preventing the synthesis of bioactive sterol and nonsterol metabolic intermediates in the cholesterol synthetic pathway. The mechanism by which these drugs cause myopathy is not clear yet.64–66 However, a recent study showed anti-HMGCR antibodies in patients with statin-induced autoimmune myopathy. It has been suggested that statins upregulate HMG-CoA autoantigen in regenerating muscle cells that in turn sustain an autoimmune response even after statin withdrawal, providing a mechanism for statin-induced immune-mediated necrotizing myopathy.67 Other drugs such as hydroxyurea can cause skin rashes that resemble DM.68 TNF inhibitors have been associated with the onset of autoimmune diseases such as vasculitis and a lupus-like syndrome. Recent reports implicate that TNF inhibitors in inflammatory arthritis patients may either induce or exacerbate DM or anti-Jo-1 positive PM.69,70 Some other reports point to the vaccine adjuvant aluminum hydroxide as a cause of macrophagic myofasciitis. The histology shows infiltration by macrophages and some CD8+ T cells into the endomysium, perimysium, and epimysium, together with clinically elevated creatine kinase (CK) levels, muscle weakness, myalgias, fatigue, and arthralgias.71 Despite some reports of vaccine-induced myositis, systematic investigation has failed to link any vaccine to myositis.72
Pathogenesis
Significant advances have been made in our understanding of the pathogenesis of the human inflammatory myopathies.73–78 It is generally thought that IIMs are autoimmune in origin because they are frequently associated with other autoimmune diseases (e.g., Hashimoto’s thyroiditis) and collagen vascular diseases (e.g., scleroderma); many patients exhibit an autoantibody response including the presence of myositis-specific autoantibodies; some studies provide evidence for lymphocyte-mediated muscle fiber injury; and a favorable response to immunosuppressive therapies in some patients supports an autoimmune cause of these disorders.
Humoral Immune Response
More than 50% of all IIM patients have uniquely defined autoantibodies—some of which are specific to myositis, and some of which are merely associated with myositis. These are generally referred to as myositis-specific autoantibodies (MSAs) and myositis-associated autoantibodies (MAAs), respectively. MAAs include autoantibodies to various nuclear and cytoplasmic antigens. Antinuclear antibodies (ANAs) present in myositis are not particularly associated with any disease subgroup, whereas MSAs that are directed against antigens of the protein synthesis pathway (e.g., aminoacyl–transfer RNA [tRNA] synthetases and signal recognition particles) and nuclear components (e.g., nuclear helicase [Mi-2]) are often associated with distinct clinical disease groups and subgroups (e.g., tRNA synthetases with interstitial lung disease, Mi-2 with DM) (Table 85-3).
Table 85-3 Myositis-Specific Antibodies
| Autoantibodies | Clinical Disease/Features |
|---|---|
| Antisynthetase autoantibodies* | More common in polymyositis than dermatomyositis; interstitial lung disease, arthritis, Raynaud’s phenomenon, fevers, mechanic’s hands |
| Signal recognition particle (SRP)† | Polymyositis; possible severe disease and cardiac involvement |
| Chromodomain helicase DNA binding proteins 3 and 4 (Mi-2α and β)‡ | Dermatomyositis |
* Common antisynthetase antibodies found in myositis are targeted to histidyl-tRNA synthetase (Jo-1), threonyl-tRNA synthetase (PL-7), alanyl-tRNA synthetase (PL-12), isoleucyl-tRNA synthetase (OJ), glycyl-tRNA synthetase (EJ), and asparaginyl-tRNA synthetase (KS).
† Autoantibodies commonly bind to a 54-kD SRP protein in the U.S. patient population and 72-, 54-, and 9-kD proteins in the Japanese population.
‡ Targets a 240-kD helicase protein that is part of the nucleosome remodeling deacetylase complex.
Anti–histidyl-tRNA synthetase antibodies are the most frequent and are present in about 16% to 20% of myositis patients.79–81 Antibodies against other aminoacyl-tRNA synthetases such as threonyl-tRNA synthetase (PL-7), alanyl-tRNA synthetase (PL-12), isoleucyl-tRNA synthetase (OJ), glycyl-tRNA synthetase (EJ), and asparaginyl-tRNA synthetase (KS) are found less frequently (1% to 3%). Anti–Mi-2 antibodies are strongly associated with DM,82,83 with prominent features such as Gottron’s papules, heliotrope rash, the V sign, and the shawl sign. An individual usually has only one MSA because MSAs are often mutually exclusive. The MSAs are most common in patients with other autoimmune diseases and are infrequent or absent in IBM patients and those with malignancies, muscular dystrophies, or other myopathies. These antibodies are sometimes present before the onset of clinical disease.84
MAAs such as PM-Scl are frequently associated with a characteristic overlap syndrome that includes features of scleroderma.85,86 This syndrome is characterized by mild muscle disease, prominent arthritis, and limited skin involvement; it frequently responds to therapy.87 Some myositis patients also have other MAAs such as anti-snRNP, anti-Ro/SSA, anti-Ku, and anti-PMS1. Antibodies recognizing an uncharacterized 56-kD large nuclear ribonucleoprotein have been found in a majority of myositis patients (86%), and the antibody titer appears to vary with disease activity, suggesting its importance in our understanding of disease pathogenesis and its potential usefulness as a clinical disease marker.88 Some of the MSAs show strong immunogenetic associations; for example, antibodies against aminoacyl-tRNA synthetases are associated with HLA-DQA1*0501, anti-SRP with DR5, anti–Mi-2 with DR7, and anti–PM-Scl with DR3.37 Neither the molecular mechanisms that initiate and perpetuate the autoimmune response nor the precise role of these autoantibodies in the pathogenesis of myositis is currently known. However, these antibodies serve as excellent clinical markers and can help diagnose and categorize these heterogeneous disorders into homogeneous subgroups.
Cell-Mediated Immune Response
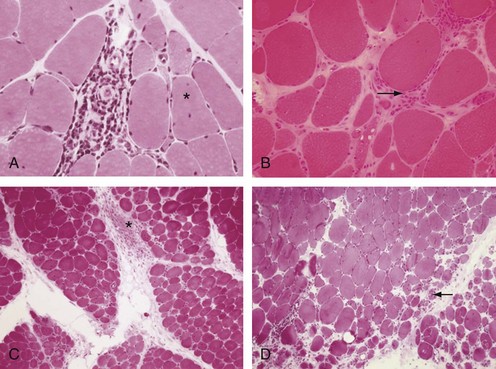
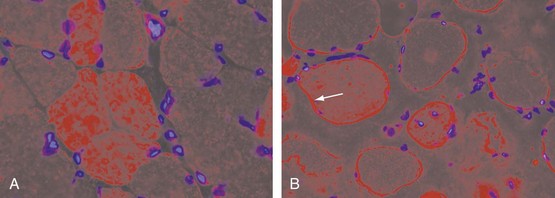
At the cellular level, there are distinct differences in the distribution and location of the various lymphocyte subsets in the muscle tissues in different IIMs. Two major patterns of inflammatory cell infiltrates are seen in muscle tissue. The first has a predominantly perivascular distribution (Figure 85-1A), often in perimysial areas (Figure 85-1C), and is largely made up of CD4+ T cells, macrophages, and dendritic cells. Occasionally, B cells are present in some patients. This pattern is seen mainly in DM patients with skin rash but occasionally in patients without a rash. The second pattern has a predominantly endomysial distribution (Figure 85-1B), with mononuclear inflammatory cells often surrounding and sometimes invading non-necrotic muscle fibers. These inflammatory cellular infiltrates are comprised primarily of CD8+ T cells and macrophages, but CD4+ T cells and dendritic cells are also present. This pattern is generally seen in patients without skin rashes and often in those classified as having PM or IBM. In some patients, the two patterns of inflammation are seen in the same biopsy. The two distinct locations and the varying compositions of the inflammatory cell populations in the two areas suggest two different pathogenic mechanisms—one that targets the blood vessels and one that targets the muscle fibers. Notable inflammation is also seen in other organs.
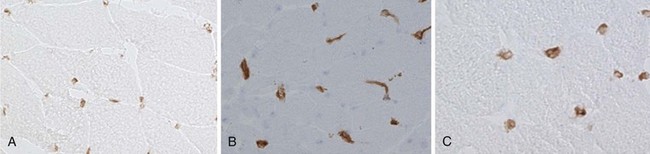
The vascular involvement in patients with DM is also manifested in the skin and can be seen clinically in the form of nail-fold changes and changes in the gastrointestinal (GI) tract. The capillaries show clear hyperplasia, vacuolization, and necrosis, contributing to an ischemia that could cause fiber damage.89,90 One of the earliest events in the pathogenesis of DM appears to be activation of the complement cascade. This leads to the subsequent deposition of complement components, which in turn results in the deposition of lytic membrane attack complexes in the endothelial cells and the eventual loss of capillaries due to complement-mediated damage. The capillaries are abnormally thickened and enlarged and look like high endothelial venules, which are characteristics of vessels that facilitate lymphocyte trafficking (Figure 85-2). The capillaries also show signs of neovascularization.91 This loss of capillaries results in some of the histopathologic features characteristic of this disease: capillary necrosis and loss, perivascular inflammation and ischemia (rarely seen), and perifascicular atrophy (a late feature; see Figure 85-1C and D). Recent studies also point out a role for type I interferon (IFN)-inducible genes in the pathogenesis of DM. It has been shown that plasmacytoid dendritic cells produce type I IFN and induce expression of IFN-inducible proteins such as MxA and IFN-inducible gene 15 (ISG15) at perifascicular myofibers and capillaries of DM biopsies, suggesting that injury to muscle fibers and capillaries occurs due to the intracellular overproduction of one or more type I IFN-inducible proteins in DM92,93-induced genes Although no direct comparison has been reported, the pathologic changes in juvenile and adult DM appear to be similar, except that all the basic pathologic features are more prominent in the childhood form (see later). The factors that initiate complement activation in this disease are poorly understood; however, the consequences of complement-mediated damage are clearly visible in DM.94
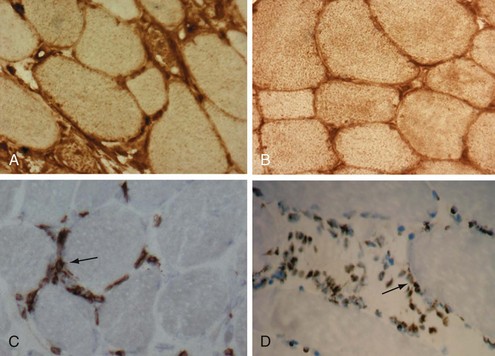
The endomysial inflammatory aggregates contain a high percentage of T cells, particularly activated CD8+ T cells, macrophages, and CD4+ T cells and few natural killer cells. Immunoelectron microscopic studies have provided evidence of the invasion, replacement, and probable destruction of non-necrotic muscle fibers by T cells and macrophages.95 It is suggested that CD8+ cytotoxic T lymphocytes (CTLs) recognize major histocompatibility complex (MHC) class I on muscle fibers and may mediate muscle fiber damage. Infiltrating CTLs express perforin-containing granules, which are characteristically oriented toward the target muscle fiber, indicating that muscle fiber injury may be partially mediated by perforin-dependent cytotoxic mechanisms (Figure 85-3B and C).96 In PM and IBM, there is evidence of clonal proliferation of CD8+ T cells, both within the muscle and in the peripheral circulation.97,98 T cell lines from patients demonstrate cytotoxicity against autologous myotubes,99 suggesting that the muscle fiber injury in PM and IBM is mediated by CTLs. CTLs are known to mediate target cell damage by both perforin–granzyme B and Fas-FasL pathways. The overexpression of antiapoptotic molecules such as Bcl-2, Fas-associated death domain–like IL-1 converting enzyme inhibitory protein (FLIP), and human inhibitor of apoptosis protein–like protein in skeletal muscle of myositis patients suggests that perforin–granzyme B–mediated CTL damage may play a predominant role in muscle fiber injury and dysfunction in myositis.100–102
In contrast to previous concepts, recent studies also show accumulations of B cells, plasma cells, myeloid dendritic cells, late-activated macrophages expressing 25F9 marker, as well as CD8+ CD28– and CD4+ CD28– T cells (TCR V[β]-expanded T cells) in the skeletal muscle and in the peripheral circulation of PM, DM, and IBM patients. These CD28– cells and late-activated macrophages expressing 25F9 marker are hypothesized to exhibit cytotoxic potential and produce proinflammatory cytokines in IIM skeletal muscle.103,104 Another study also explored the potential role of FOXP3+ Treg cells in the pathology of myositis. These authors reported that the number of Treg cells correlated with the degree of inflammation in IIMs and suggested that these cells might serve to counterbalance activity of cytotoxic T cells in myositis.105
On the basis of the data described, two different pathways have been proposed as major mediators of muscle damage and inflammation: one mediated through T lymphocytes (CTLs) directed against muscle fibers, predominating in PM and IBM, and the other directed against vessels, predominating in DM. However, several studies have shown that the degree of inflammation does not consistently correlate with the severity of the structural changes in the muscle fibers or with the severity of the clinical disease,106 suggesting that nonimmune processes also play a role in disease pathogenesis. A role for nonimmune processes is supported by the following observations: First, marked structural changes in the muscle fibers occur in the absence of any inflammatory cells.107,108 Second, there is a lack of correlation between the degree of inflammation and the degree of muscle weakness.109 Third, some myositis patients do not respond, even to powerful anti-inflammatory therapy.110,111 Fourth, steroid treatment may eliminate inflammatory cells in myositis muscle tissue, but this removal alone may not substantially improve the clinical disease, suggesting that immunosuppressive therapies modulate disease activity but do not change other mediators of the disease process.112 Finally, the clinical disease may progress when identifiable inflammation has subsided,113 suggesting a role for nonimmune mechanisms in the pathogenesis of myositis. Thus the exact contribution of immune-mediated pathways to muscle damage is currently unknown.
Class I Major Histocompatibility Complex
Normal skeletal muscle cells do not constitutively express or display MHC class I molecules, although they can be induced to do so by proinflammatory cytokines such as IFN-γ or TNF107,114–116 or by the alarmin HMGB1.139 In contrast, in human IIMs, the early and widespread appearance of MHC class I in non-necrotic muscle cells is a striking feature, even in muscle cells distant from the lymphocytic infiltration.107,108,117 MHC class I staining is usually observed on the sarcolemma of muscle fibers, but some fibers also show staining in both the sarcolemma and the sarcoplasm (see Figure 85-3A and B). In some patients, the expression is restricted to a few clusters (often in early disease), whereas in others, almost every fiber is positively stained, particularly in late-phase and treatment-resistant cases. The biologic significance of these observations has been explored by generating a conditional transgenic mouse model overexpressing syngenic mouse MHC class I. The overexpression of MHC class I molecules in the skeletal muscle of mice results in the development of clinical, biochemical, histologic, and immunologic features that resemble human myositis and provides a close model of the human disease. The disease in these mice is inflammatory, limited to skeletal muscles, self-sustaining, more severe in females, and often accompanied by MSAs.118 Recent studies in this model further suggest that MHC class I overexpression leads to endoplasmic reticulum stress, muscle atrophy, and decrease in force generation capacity of skeletal muscle, implicating the role for MHC class I muscle weakness in myositis.119,120
A number of observations in human myositis patients and in the mouse model of myositis suggest that MHC class I molecules themselves may mediate muscle fiber damage and dysfunction in the absence of lymphocytes. For instance, in human myositis, the induction of MHC class I antigen in muscle fibers occurs early, preceding inflammatory cell infiltration.121,122 MHC class I staining of human myositis biopsies shows both a cell surface and a sarcoplasmic reticulum pattern of internal reactivity, demonstrating that some of the MHC class I molecules may be retained in the endoplasmic reticulum (ER) of these fibers.78,108,123 Persistent MHC class I overexpression in muscle fibers may exist in the absence of an inflammatory infiltrate.113 The controlled induction of MHC class I in the mouse model is followed by muscle weakness before mononuclear cell infiltration.118 It has recently been shown that in vivo gene transfer of MHC class I plasmids attenuates muscle regeneration and differentiation.124 Together, these observations, and particularly the obvious retention of MHC class I within the cell in both human and murine disease, indicate that the muscle fiber damage seen in myositis may not be solely mediated by immune attack (e.g., CTLs, autoantibodies); it may also be mediated through nonimmunologic mechanisms such as the ER stress response and hypoxia.
Because MHC class I assembly occurs in the ER and because upregulation in myositis muscle fibers is widespread, even in the absence of visible inflammatory infiltrate, it is likely that ER stress plays a role in the muscle fiber damage and dysfunction associated with human myositis. The ER is intimately involved in the folding, exporting, and processing of newly synthesized proteins. When there is an imbalance between the protein load in the ER and the cell’s ability to process that load, a series of signaling pathways that adapt cells to ER stress is activated. This ER stress response can be provoked by a variety of pathophysiologic conditions including ischemia, hyperhomocysteinemia, viral infections, and mutations that impair protein folding, as well as by excess accumulation of protein in the ER.125,126 Cells self-protect against ER stress by initiating at least four functionally distinct responses: (1) upregulation of the nuclear factor κB (NFκB) pathway (ER overload response); (2) upregulation of genes encoding ER chaperone proteins such as Bip/GRP78 and GRP94, as a means of increasing protein folding activity and preventing protein aggregation; (3) translational attenuation to reduce the load of protein synthesis and to prevent the further accumulation of unfolded proteins (unfolded protein response); and (4) cell death, which occurs when the ER’s functions are severely impaired. This cell death event is mediated by transcriptional activation of the gene for CHOP/GADD153, a member of the C/EBP family of transcription factors,127 and by the activation of ER-associated caspase 12.128
In myositis, it appears that overexpression of MHC class I in myofibers initiates a series of cell autonomous changes that contribute to myofiber pathology. Recent investigations have indicated that overexpression of MHC class I on muscle fibers results in activation of the NFκB and ER stress response pathway in human inflammatory myopathies and in the mouse model of myositis.78,129 NFκB can be activated within minutes by a variety of stimuli including inflammatory cytokines such as TNF and IL-1, T cell activation signals, and stress inducers. It is likely that in human myositis, NFκB activates both classic (proinflammatory cytokines) and nonclassic (ER stress response) pathways.78,129–132 Further, there is evidence that downstream target genes (e.g., MHC class I, intercellular adhesion molecule [ICAM], monocyte chemoattractant protein [MCP]-1) regulated by the NFκB pathway are highly upregulated in myositis patients.123,133,134 Recent studies have indicated that NFκB p65 is activated both in human myositis biopsies and in the mouse model,78,129,135,136 suggesting that this pathway may be directly involved in muscle fiber damage (Figure 85-4). NFκB is a potential therapeutic target in myositis, and the use of NFκB pathway inhibitors significantly reduces the pathology associated with several autoimmune diseases including diabetes, multiple sclerosis, inflammatory bowel disease, and rheumatoid arthritis, suggesting that this pathway is a critical player in the effector phase of autoimmune pathology. Thus it appears that MHC class I expression on muscle fibers links the immune and nonimmune mechanisms of muscle fiber damage.
Cytokines and Hypoxia
A number of other effector molecules produced in muscle tissue by inflammatory cells, endothelial cells, and muscle fibers are thought to play a role in the pathogenesis of myositis.75 Most of the data assembled relate to cytokines, but some data related to chemokines are also available. The most consistently demonstrated cytokines in muscle tissue from patients with IIMs are cytokines with proinflammatory properties: IL-1α, IL-1β, TNF, and IFN-α. Recently, IL-10, IL-13, epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), CCL3 (macrophage inflammatory protein [MIP-1α]), CCL4 (MIP-1β) and CCL11 (eotaxin), IL-15, and IL-15Rα were also demonstrated to be significantly upregulated and granulocyte colony-stimulating factor (G-CSF) downregulated in patients with IIMs relative to normal subjects.137,138 Further, the DNA-binding high mobility group box 1 (HMGB1) was found to exhibit both extranuclear and extracellular patterns in the muscle tissue of patients with PM and DM. Stimulation with IFN-γ showed an increased HMGB1 expression in muscle nuclei and the myoplasm. Exposure to HMGB1 induced a reversible upregulation of MHC class I in the muscle fibers and irreversible decrease in Ca2+ release from the sarcoplasmic reticulum during fatigue, implicating a role of HMGB1 and MHC class I early in the pathogenesis of IIMs.139 In addition to inducing the upregulation of MHC class I and II molecules on muscle fibers, cytokines may have a direct effect on muscle fiber function, as has been demonstrated for TNF.140 The relative importance of the various cytokines and chemokines in patients with myositis is still uncertain, but these molecules are potential biomarker candidates in this disease as exemplified by a recent study showing serum IL-6 production and the type I IFN gene signature in the peripheral blood correlating with disease activity in DM patients.141
Microvessel involvement was first observed in DM but has also become evident in PM. The endothelial cells in both subsets show increased expression of adhesion molecules and proinflammatory cytokines such as IL-1α. This phenotype can be induced by tissue hypoxia, which may result from capillary loss and local tissue inflammation. Muscle tissue hypoxia can contribute to the clinical symptoms and muscle fatigue and might be associated with disease mechanisms in inflammatory myopathies.75 A recent study reported the expression of VEGF receptor in muscle fibers and HIF-2α reactivity in endothelial cells of PM and IBM patients DM patients showed hypoxia-inducible factor (HIF)-1α and HIF-1β expression in endothelial cells, whereas expression of HIF-2α, erythropoietin receptor, VEGF, and VEGF-R were also observed on muscle fiber. These observations suggest that deprivation of blood supply by immune-mediated mechanisms might trigger the upregulation of hypoxia-related proteins as an adaptive response.142 The hypoxia hypothesis is further supported by the clinical improvement observed after exercise, but a causal connection still needs to be established. In addition, magnetic resonance spectroscopic analysis, before and after a work load, has demonstrated reduced levels of energy substrates that are important for muscle contraction such as adenosine triphosphate and phosphocreatine, when compared with levels in healthy individuals. This finding supports the hypothesis that an acquired metabolic disturbance occurs in chronic inflammatory myopathies and that this disturbance can contribute to impaired muscle performance.
Proposed Mechanisms of Muscle Damage
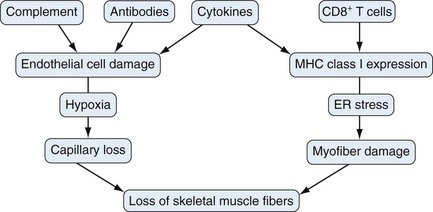
Currently available data suggest that both immune (cell-mediated and humoral) and nonimmune (ER stress, hypoxia) mechanisms play a role in muscle fiber damage and dysfunction in myositis. ER stress, hypoxia, and the NFκB pathway are highly active within the skeletal muscle of myositis patients, and the proinflammatory NFκB pathway connects the immune and nonimmune components contributing to muscle damage. The relative contribution of each of these pathways to muscle fiber damage is presently unclear (Figure 85-5). Therefore use of specific drugs to inhibit these pathways, either alone or in combination, would help define their roles in myositis and potentially serve as effective therapeutic agents.
Clinical Features
The inflammatory myopathies may occur as distinct disease entities, or they may coexist with some other rheumatic disease. This observation is true for all three subsets of myositis, but it is most often seen in PM and DM. The rheumatic diseases most often associated with inflammatory myopathies are systemic sclerosis, mixed connective tissue disease, Sjögren’s syndrome, and SLE; however, rheumatoid arthritis may also be associated with inflammatory myopathies. IBM may be associated with Sjögren’s syndrome, SLE, and other autoimmune diseases.143,144 Because the clinical features of IBM differ somewhat from those of PM and DM, they are presented separately.
Polymyositis and Dermatomyositis
Skin
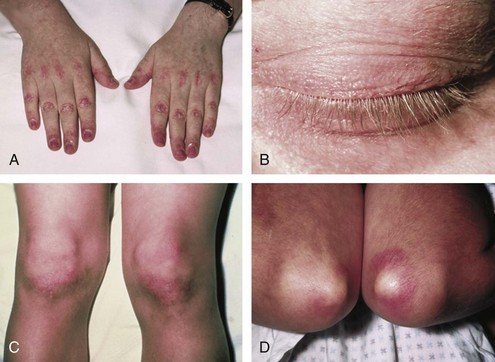
Dermatomyositis is characterized by the presence of certain types of rashes145; the same types are often seen in both children and adults. The most specific skin manifestations are Gottron’s papules and the heliotrope rash (Figure 85-6). Gottron’s papules are slightly elevated violaceous, pink, or dusky red papules located over the dorsal side of the metacarpal or interphalangeal joints. These papules may also occur over the extensor side of the wrist, elbow, or knee joints. Gottron’s papules are considered to be pathognomonic of DM. A macular rash (without papules) with the same distribution as Gottron’s papules is called Gottron’s sign (see Figure 85-6C and D). The heliotrope rash is a periorbital red or violaceous erythema of one or both eyelids, often with edema (see Figure 85-6B). Linear erythema overlying the extensor surfaces of joints is also relatively specific to DM (Figure 85-7A). Many patients with DM have photosensitive rashes, typically located on the face or scalp or over the neck (the so-called V sign), although this rash is not specific to DM (Figure 85-7B and C). Another common rash in DM is located over the shoulders (shawl sign; Figure 85-7D) or over the hips (holster sign). Pruritus is common. Patients with DM often have skin lesions on their fingers such as periungual erythema, nail-fold telangiectasias, and cuticular overgrowth (Figure 85-8C). Other less common skin manifestations are panniculitis, livedo reticularis, and nonscarring alopecia. Vasculitis may be seen in children with DM but rarely in adults.
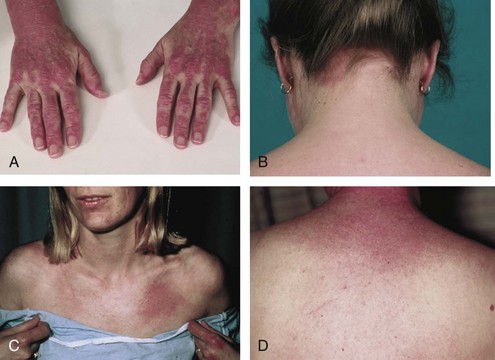
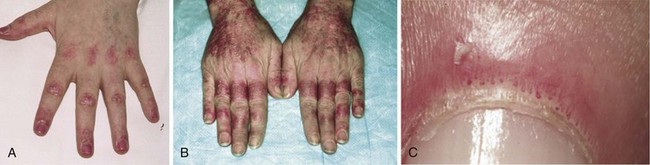
In general, the skin rash is moderate, with local erythema. In rare cases, a severe, diffuse erythema (erythroderma) may occur, occasionally with vesiculobullous lesions or ulcers. The skin rash may precede the muscle symptoms by months or even years, and in some patients, the skin manifestations may be the only clinical sign of DM; this condition is often called amyopathic DM or DM sine myositis (see later). The pattern of the rash over the knuckles and dorsum of the hand is distinct, in that the rash generally affects the phalanges but spares knuckles in SLE, and vice versa in DM (Figure 85-8A and B). However, no histopathologic skin features are specific for DM; most of the features are also seen in patients with SLE. Thus skin biopsy is rarely helpful in distinguishing between these two disorders. The cutaneous manifestations may fail to respond to immunosuppressive treatment, despite improvement in muscle symptoms. Thus it is possible that different molecular pathways or disease mechanisms cause the skin rash and the muscle inflammation.
Calcinosis, which can be severe, is found mainly in juvenile DM but is occasionally seen in adults. The calcinosis occurs predominantly in sites that have been subject to friction or trauma such as the elbows or knees. Sometimes the calcinosis can be extensive and erupt, leading to ulcers. It is most often localized to the subcutaneous tissue but can also develop in the skin, fascia, or muscle and can be visualized by radiography, computed tomography (CT), or magnetic resonance imaging (MRI). The calcinosis seems to progress as long as there is active inflammatory disease. Also, once it has developed, it is often treatment resistant. Some data, however, suggest that the progress of calcinosis can be inhibited by effectively treating the inflammatory process in the skin and muscle.146
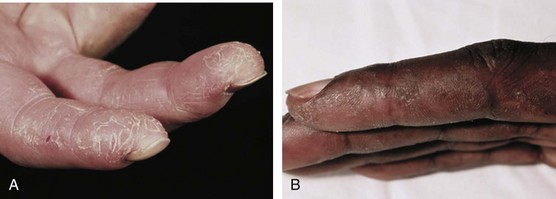
Another type of skin pathology seen in inflammatory myopathies is called mechanic’s hands. This rash is often associated with the presence of antisynthetase autoantibodies and can be seen in both PM and DM. The rash is a hyperkeratotic, scaling, fissuring of the fingers, particularly on the radial side of the index fingers (Figure 85-9).
Lungs
Lung involvement is frequent in PM and DM and is a major risk factor for morbidity and mortality. Clinical symptoms such as dyspnea and cough are common. Lung involvement can be caused by weakness of the respiratory muscles or inflammation of the lung tissue (interstitial lung disease). Weakness of the respiratory muscles may lead to restrictive lung disease, and involvement of the pharyngeal muscles is a risk factor for aspiration pneumonia. Interstitial lung disease, caused by inflammation in the small airways, is common in PM and DM and is often associated with antisynthetase autoantibodies; it may be present in up to 70% of patients when investigated with sensitive techniques such as high-resolution CT and measurement of pulmonary function and diffusion capacity.147 In most cases, the changes are present at the time of diagnosis of myositis; they rarely develop after immunosuppressive treatment has started. The severity of interstitial lung disease may vary from mild or even asymptomatic to rapidly progressive (Hamman-Rich-like) with a fatal outcome. In most cases, the interstitial lung disease is mild and has a slowly progressive course. In some cases, improvement in lung function is seen with immunosuppressive treatment. The course and outcome vary, depending on the histopathology, suggesting that different disease mechanisms cause interstitial lung disease.
Antisynthetase Syndrome
A new classification system is based on the presence of MSAs, rather than clinical and histopathologic changes. The most common of these antibodies are the antisynthetase autoantibodies directed against aminoacyl-tRNA synthetases. A clinically distinct subset of myositis, often called antisynthetase syndrome, has been identified in patients with antisynthetase autoantibodies.42,63 The most common of the antisynthetase autoantibodies is anti–Jo-1, which is directed against histidyl-tRNA synthetase. This autoantibody is present in approximately 20% of patients with PM or DM but is only rarely found in patients with IBM.79 Antisynthetase syndrome is characterized by the presence of antisynthetase autoantibodies and a set of clinical features that includes myositis, interstitial lung disease, Raynaud’s phenomenon, nonerosive symmetric polyarthritis of the small joints, and mechanic’s hands (see Figure 85-9). These patients often have fever at disease onset and during flares of disease. Antisynthetase syndrome can be seen in patients with PM or DM but is more often seen in patients without skin rashes other than mechanic’s hands.
Amyopathic Dermatomyositis
A subset of DM is called clinically amyopathic DM. These patients have a skin rash, which is typical of DM, but no clinical signs of muscle involvement.148 The proposed definition is based on a skin biopsy consistent with DM and a duration of 6 months or longer in the absence of clinical or laboratory evidence of myositis. Some of these patients do have subclinical myositis based on MRI or biopsy findings at presentation; others develop clinically overt myositis sometime later. Patients without clinically overt myositis, however, may develop extramuscular manifestations such as interstitial lung disease, which may be severe. Amyopathic DM may be associated with malignancies, as is the case for classic DM. The frequency of this subset is uncertain, but some recent studies suggest that this form of DM may be more common than previously thought.
Juvenile Dermatomyositis
The incidence of juvenile dermatomyositis (JDM) is between 1.7 and 3 per million children. The disease onset has two peaks—at age 6 and 11 years. JDM is more common in girls than in boys in Europe and North America; in Japan and Saudi Arabia, this difference is less prominent. The most common clinical manifestations at disease onset are muscle weakness, easy fatigability, skin rash, malaise, and in some cases fever.146 The skin rash is often pathognomonic and similar to adult DM, with the most typical skin manifestation being heliotrope discoloration of the upper eyelids, Gottron’s papules, periungual erythema, and capillary loop abnormalities. Calcinosis, cutaneous ulceration, and lipodystrophy are more common in juvenile cases than in adults. Calcinosis is seen in 30% to 70% of children with JDM. The calcinosis is most often located at sites exposed to trauma and can be seen in the skin, fascia, or muscles. In some children, the calcinosis becomes prominent and causes contractures and ulcerations. Lipodystrophy occasionally develops, and other metabolic abnormalities such as insulin resistance and hepatomegaly are sometimes seen. Vasculopathy that affects the GI tract with ulceration, perforation, or hemorrhage is rare but seems to be more common in children than in adults with DM. Because this can be a serious sign, screening for GI involvement should be included in the evaluation of patients with JDM. Interstitial lung disease is rarely seen in JDM cases.
Inclusion Body Myositis
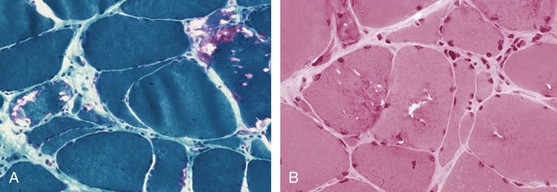
IBM is distinguished from PM and DM on the basis of both clinical and histopathologic features.149,150 Sporadic IBM is a distinct entity from familial hereditary inclusion body myopathy, which shares some clinical and histopathologic features but lacks signs of inflammation in muscle tissue. IBM was identified in the 1960s as a subset of inflammatory myopathies, distinct from PM, primarily on the basis of typical histopathologic features that include sarcoplasmic and nuclear inclusions and rimmed vacuoles.16,17 A characteristic clinical phenotype was later identified, characterized by an insidious onset of muscle weakness over months to years, muscle weakness localized predominantly to the thigh muscles and finger flexors, and resistance to glucocorticoid treatment. IBM patients often have a history of frequent falling. Sporadic IBM cases are sometimes misdiagnosed as PM because the classic histopathologic changes (rimmed vacuoles and inclusions) may not be evident in early biopsies (Figure 85-10). A slowly progressive clinical course, development of severe muscle atrophy in the thighs and forearms, and resistance to treatment with immunosuppressive drugs should raise the suspicion of IBM, and a second muscle biopsy should be considered.
< div class='tao-gold-member'>





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


