62 Immunosuppressive Drugs
Immunosuppressive drugs comprise different classes of drugs that dampen the immune system—notably T and B lymphocytes—functionally and/or numerically (Table 62-1) but do not permanently correct the fundamental imbalance of immune regulation in autoimmune disease. As such, they do not have curative potential yet they can be effective in remission induction and control of specific rheumatic disease manifestations and remain cornerstone drugs in the management of rheumatic conditions. Many immunosuppressive drugs have withstood the test of time, as attested by their ongoing use in transplantation medicine, nephrology, gastroenterology, ophthalmology, dermatology, and rheumatology. Consequently, their therapeutic potential and toxicity profiles hold few surprises. Apart from drug-specific toxicities, the main risk of immunosuppressive treatment is infection. In the absence of validated biomarkers of infection, sound clinical judgment and experience remain indispensable in monitoring patients who use immunosuppressive drugs, often for long periods of time. The use of live vaccines is contraindicated, and although other vaccinations are generally less effective, annual influenza vaccination is recommended in patients taking immunosuppressive medication.
Table 62-1 Mechanisms of Action of Immunosuppressive Drugs
| Drugs | Class | Mechanism of Action |
|---|---|---|
| Cyclophosphamide, chlorambucil | Alkylating cytotoxics | Active metabolites alkylate DNA |
| Azathioprine, mercaptopurine | Purine analogue cytotoxics | Inhibit purine synthesis |
| Cyclosporine, tacrolimus (FK506) | Calcineurin inhibitors | Inhibit calcium-dependent T cell activation and interleukin-2 production |
| Sirolimus (rapamycin) | Noncalcineurin-binding macrolide immunoregulator | Blocks interleukin-2-mediated and growth factor–mediated signal transduction |
| Mycophenolate mofetil | Purine synthesis inhibitor | Mycophenolic acid inhibits inosine monophosphate dehydrogenase |
| Thalidomide | Glutamic acid derivative | Inhibition of tumor necrosis factor production and angiogenesis |
Alkylating Agents
Cyclophosphamide
Structure
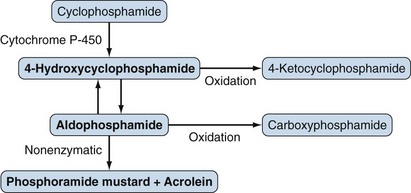
Cyclophosphamide is an oxazaphosphorine-substituted nitrogen mustard and inactive prodrug requiring enzymatic bioactivation (Figure 62-1). Cyclophosphamide is the alkylating agent of choice for most rheumatic disease requiring such therapy.
Mechanisms of Action
Its DNA-alkylating effects are mediated predominantly through phosphoramide mustard and, to a lesser extent, other active metabolites. These positively charged, reactive intermediates alkylate nucleophilic bases, resulting in the cross-linking of DNA and of DNA proteins, breaks in DNA, and consequently decreased DNA synthesis and apoptosis.1 The cytotoxicity of alkylating agents correlates with the amount of DNA cross-linking, but the relationship between cytotoxicity and immunosuppressive effects is unclear. The effects of cyclophosphamide are not exclusively limited to proliferating cells or particular cell types. Sensitivity varies among cell populations, however; for example, hematopoietic progenitor cells are relatively resistant to even high doses of cyclophosphamide. The immunosuppressive effects of cyclophosphamide include decreased numbers of T lymphocytes and B lymphocytes, decreased lymphocyte proliferation, decreased antibody production, and suppression of delayed hypersensitivity to new antigens with relative preservation of established delayed hypersensitivity.2
Pharmacology
Absorption and Distribution
Oral and intravenous (IV) administration of cyclophosphamide results in similar plasma concentrations.3 Peak plasma concentrations of cyclophosphamide occur 1 hour after oral administration. Protein binding of cyclophosphamide is low (20%), and it is widely distributed.1
Metabolism and Elimination
Cyclophosphamide is rapidly metabolized, largely by the liver, to active and inactive metabolites. The formation of the active 4-hydroxycyclophosphamide is mediated by various cytochrome P-450 (CYP) enzymes, and genetic variations in the enzymes in lupus nephritis patients have been shown to affect responses to cyclophosphamide.4 4-Hydroxycyclophosphamide, which is not cytotoxic at physiologic pH, readily diffuses into cells and spontaneously decomposes into the active phosphoramide mustard. The elimination half-life of cyclophosphamide is 5 to 9 hours, and alkylating activity is undetectable in the plasma of most patients 24 hours after a dose of 12 mg/kg.1 Plasma concentrations of cyclophosphamide are not clinically useful predictors of either efficacy or toxicity. Between 30% and 60% of the total cyclophosphamide is eliminated in the urine, mostly as inactive metabolites, although some cyclophosphamide and active metabolites such as phosphoramide mustard and acrolein can also be detected in urine.1
Pharmacokinetic Considerations in Special Circumstances
Liver Disease
Although the half-life of cyclophosphamide is increased to 12 hours in patients with liver failure compared with 8 hours in controls, toxicity is not increased, suggesting that exposure to cytotoxic metabolites is not increased and dose modification in liver disease is generally not required.1
Renal Impairment
Some studies have shown little alteration in drug disposition with no increased toxicity in patients with impaired renal function.1 In patients with autoimmune disease and a creatinine clearance of 25 to 50 mL/min and 10 to 25 mL/min, exposure to cyclophosphamide increased approximately 40% and 70%, respectively.5 In clinical practice, initial cyclophosphamide doses are therefore decreased by approximately 30% in patients with moderate-to-severe renal impairment and subsequent doses are titrated according to clinical response and effects on the leukocyte (white blood cell) count. Cyclophosphamide is removed by dialysis and is administered after dialysis, or, alternatively, dialysis can be initiated the day after cyclophosphamide administration.5
Clinical Indications
Cyclophosphamide remains the drug of choice for most patients with systemic necrotizing vasculitis or Goodpasture’s syndrome, for many patients with organ-threatening SLE, and for some patients with autoimmune disease–associated interstitial lung disease and inflammatory eye disease. In rheumatoid arthritis (RA) unless complicated by vasculitis, less toxic and more effective drugs have replaced cyclophosphamide. In SLE a remission induction course with IV cyclophosphamide followed by maintenance with azathioprine or mycophenolate to minimize cyclophosphamide toxicity is the most commonly used treatment for severe organ involvement including lupus nephritis, although remission induction regimens with MMF have been propagated as an effective and safe alternative for cyclophosphamide (discussed later). The original National Institutes of Health (NIH) protocol involved 6 monthly IV infusions with cyclophosphamide 1 g/m2 then once every 3 months for at least 24 additional months,6 whereas the Euro-Lupus protocol used in Europe involved administration of six IV infusions of 500 mg of cyclophosphamide every 2 weeks followed by azathioprine maintenance (Table 62-2). A comparison with 6 monthly IV infusions with cyclophosphamide 500 mg/m2, followed by two further infusions of slightly higher doses 3 and 6 months later, and azathioprine maintenance therapy resulted in similar rates of the end points of end-stage renal disease or doubling of creatinine concentration with up to 10 years of follow-up.7 Cyclophosphamide as either IV pulse therapy or orally can also be effective in patients with other serious complications of SLE including central nervous system involvement and thrombocytopenia and interstitial lung disease associated with systemic sclerosis and other autoimmune diseases.8–10 Several trials have investigated whether IV cyclophosphamide is as effective as oral cyclophosphamide as remission induction therapy for granulomatosus with polyangiitis (GPA), the newly proposed name for Wegener’s granulomatosis.11 Although early trial results suggested superiority of oral dosing, more recent clinical trial data pointed to equal efficacy, but slightly less hematologic toxicity with IV therapy.12–14 As in lupus nephritis, shorter induction courses of cyclophosphamide have been reported to be effective in GPA and microscopic polyangiitis.15 Cyclophosphamide has a steep dose-response curve, making it an ideal compound for dose escalation. High doses of cyclophosphamide, with or without stem cell rescue and lymphoablative antibodies or total body irradiation, have been used for severe juvenile idiopathic arthritis (JIA), RA, systemic sclerosis, and SLE.16 With the introduction of effective biologics and new treatment paradigms for RA and JIA, the clinical need for immunoablative treatment in these diseases has waned. Although large series have shown promising results of immunoablative therapy and stem cell rescue in patients with severe SLE, a recent randomized trial showed that standard-dose IV cyclophosphamide was not inferior to high-dose cyclophosphamide without stem cell rescue or lymphoablative antibodies.17 Prospective, randomized trials are in progress in systemic sclerosis to compare safety and efficacy of IV pulse cyclophosphamide and immunoablative therapy with stem cell rescue.
Table 62-2 Lupus Nephritis Treatment Protocols
IV, intravenous.
Dosage and Route of Administration
Typical dosage regimens are presented in Table 62-2. Dosages for IV pulse therapy with cyclophosphamide range from 0.5 to 1 g/m2 and for oral therapy 2 mg/kg. The bioavailability of oral cyclophosphamide is excellent.
Toxicity
Hematologic.
Reversible myelosuppression manifesting as leukopenia and neutropenia is common and dose dependent. Generally, platelet counts are not affected with IV pulse doses of less than 50 mg/kg, but with long-term oral use, a mild decrease in platelet count is common. After a single IV dose of cyclophosphamide, the approximate times to nadir and recovery of leukocyte counts are 8 to 14 days and 21 days, respectively.18 The white blood cell nadir is about 3000 cells/mm3 after a dose of 1 g/m2 (≈25 mg/kg) and 1500 cells/mm3 after a dose of 1.5 g/m2. With long-term use, there is increased sensitivity to the myelosuppressive effects of cyclophosphamide and doses usually need to be decreased over time.
Infection
Infection with a range of common and opportunistic pathogens is a frequent complication. In 100 patients with SLE, infection occurred in 45 patients during treatment with a cyclophosphamide-based regimen and was the primary cause of death in 7 patients.19 In this study, infection was equally common in patients receiving oral or IV cyclophosphamide and was associated with a white blood cell nadir at some point in treatment of less than 3000/mm3 (55% infection rate vs. 36%). At the time of infection, the average white blood cell count was normal, however.19 A higher maximal corticosteroid dose was also associated with increased risk of infection. Half of the infections occurred at prednisone doses of less than 40 mg/day, and a quarter of the infections occurred at doses less than 25 mg/day. Lower rates of infection (25% to 30%) have been reported in SLE patients receiving cyclophosphamide in National Institutes of Health (NIH) protocols.20 Oral cyclophosphamide regimens generally pose a greater risk of infection than IV pulse regimens. Serious infections occurred in 41% and 70% of patients with GPA treated with pulse IV and daily oral cyclophosphamide, respectively.12 These rates of infection are higher than rates reported in long-term NIH protocols, in which 48% of 158 patients experienced 140 infections requiring hospitalization.21 The reported frequency of cyclophosphamide-associated infection varies, probably as a function of the stage and severity of the underlying disease, the degree of cyclophosphamide-induced immunosuppression, and variations in concomitant glucocorticoid regimens. Pneumocystis jiroveci pneumonia has been recognized as a preventable, serious opportunistic infection that complicates treatment of systemic vasculitis with regimens using cyclophosphamide and methotrexate. The risk is highest during the remission induction phase and is greater with oral than IV cyclophosphamide regimens.22 Surprisingly, in two placebo-controlled, randomized clinical trials in scleroderma lung disease, active treatment for 1 year with either oral cyclophosphamide or sequential treatment with prednisolone plus IV cyclophosphamide followed by azathioprine was not associated with more toxicity, however, suggesting disease-specific differences in toxicity.9,10
Urologic
The bladder toxicities of cyclophosphamide, hemorrhagic cystitis, and bladder cancer are related to route of administration, duration of therapy, and cumulative cyclophosphamide dose. Bladder toxicity, a particular problem with long-term oral cyclophosphamide, is largely due to acrolein, a metabolite of cyclophosphamide. It is commonly accepted that bladder toxicity can be minimized in patients receiving pulse doses of IV cyclophosphamide by administering mesna, a sulfhydryl compound that binds acrolein in the urine and inactivates it.23 Direct evidence for the effectiveness of mesna in preventing cystitis, however, comes from its use with ifosfamide in patients with cancer and data from animal models. The data from rheumatology series are consistent with a protective effect but are inadequate to come to firm conclusions, which explains differences between national guidelines.24 The short half-life of mesna renders it suboptimal for the prevention of bladder toxicity in patients receiving daily oral cyclophosphamide—but oral mesna administered three times a day with daily oral cyclophosphamide decreased the incidence of bladder toxicity to 12%.25
Nonglomerular hematuria, which may range from minor, microscopic blood loss to severe, macroscopic bleeding, is the most common manifestation of cyclophosphamide-induced cystitis.26 Nonglomerular hematuria occurred at some time in 50% of 145 patients treated with oral cyclophosphamide and was related to the duration of therapy and cumulative cyclophosphamide dose.26 The risk of bladder cancer was increased 31-fold (95% confidence interval [CI], 13-fold to 65-fold), and 7 patients (5%) had developed bladder cancer anytime between 7 months and 15 years after initiating therapy. The cancer was preceded by nonglomerular hematuria in all patients. Six of the seven patients had a cumulative dose of more than 100 g of cyclophosphamide and a duration of therapy of more than 2.7 years. Smokers were at increased risk of hemorrhagic cystitis and bladder cancer.
Malignancy
Cyclophosphamide increases the risk of malignancies (other than bladder cancer) twofold to fourfold. In the largest study, 119 patients with RA who had been treated with oral cyclophosphamide were followed for 20 years.26 There were 50 cancers in 37 patients in the cyclophosphamide group compared with 26 cancers in 25 of 119 control RA patients. Bladder, skin, myeloproliferative, and oropharyngeal malignancies occurred more commonly in the cyclophosphamide group. The risk of malignancies increased with the cumulative dose of cyclophosphamide, and 53% of patients who received more than 80 g of cyclophosphamide developed malignancy. Few malignancies have been reported in patients treated with pulse IV cyclophosphamide regimens. Current data do not allow quantification of the long-term risk of malignancy associated with pulse IV cyclophosphamide treatment, but it is likely to be substantially smaller than that associated with oral regimens.
Reproductive
Cyclophosphamide, as used in autoimmune disease, results in significant gonadal toxicity. The risk of sustained amenorrhea after cyclophosphamide therapy has ranged from 11% to 59%.27 The risk of ovarian failure depends more on age of the patient and cumulative dose of cyclophosphamide than on route of administration.27 Patients younger than 25 years old receiving 6 pulses of IV cyclophosphamide had a low frequency of ovarian failure (none of four patients), whereas patients older than 31 years receiving 15 to 24 pulses all had ovarian failure (four of four patients). The use of alkylating agents in male patients leads to azoospermia, and, if the clinical situation allows, referral to a fertility clinic for banking of sperm (or ova in female patients) should be considered before cyclophosphamide treatment. There was no increase in genetic disease in the offspring of adults who underwent cancer chemotherapy in childhood.28
Pulmonary
Cyclophosphamide-induced pulmonary toxicity occurs in less than 1% of patients. Early-onset pneumonitis 1 to 6 months after exposure to cyclophosphamide may respond to withdrawal of the drug and treatment with corticosteroids. A more insidious, irreversible, late-onset pneumonitis and fibrosis with radiographic findings of diffuse reticular or reticulonodular infiltrates may occur after treatment with oral cyclophosphamide for 1 to 13 years.29
Miscellaneous
A varying degree of reversible alopecia can occur with daily oral and monthly pulse cyclophosphamide. Cardiotoxicity, a dose-limiting adverse effect in oncology, and water intoxication, owing to inappropriate antidiuretic hormone secretion, are rare at standard doses.30 Unusual hypersensitivity reactions include urticaria and anaphylaxis, although the bladder protectant mesna is a more likely cause of allergic responses in patients receiving both drugs.31,32
Strategies to Minimize Toxicity
Strategies to minimize toxicity include adjusting the dose of cyclophosphamide to avoid a significant degree of leukopenia (white blood cell count <3000/mm3 for daily oral therapy or a nadir of <2000/mm3 for pulse IV therapy) and granulocytopenia.33 The blood count is monitored initially at 1- to 2-week intervals and monthly thereafter in patients on stable oral doses. To decrease the risk of infection added by concomitant high-dose corticosteroids, the dose of corticosteroids should be reduced after a clinical response has been obtained. Alternate-day glucocorticoids can be considered in the maintenance phase. Oral cyclophosphamide is best administered as a single dose in the morning with the patient drinking plenty of fluids and emptying the bladder frequently to dilute the urinary concentration of acrolein and to minimize the time the bladder is exposed to it. Prophylaxis against P. jiroveci pneumonia is often prescribed, particularly during the induction phase when doses of cyclophosphamide and corticosteroids are higher. The use of mesna to prevent bladder toxicity is described earlier. Urinalysis should be performed monthly, and nonglomerular hematuria should be evaluated by a urologist. All patients who receive cyclophosphamide, particularly patients who develop hemorrhagic cystitis, are at increased risk of developing bladder cancer, and lifelong surveillance is required with urinalysis, urine cytology, and, if indicated, cystoscopy.34 Lastly, drugs that are less toxic than cyclophosphamide such as methotrexate and MMF are an option for inducing remission or remission maintenance in patients with GPA or lupus nephritis.35,36
Pregnancy and Lactation
Cyclophosphamide is a U.S. Food and Drug Administration (FDA) Pregnancy Category D drug. Cyclophosphamide is teratogenic, particularly in the first trimester, and should be avoided in pregnancy and during lactation.37,38 If a patient becomes pregnant while taking (receiving) this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
Drug Interactions
Cimetidine inhibits the activity of several hepatic enzymes. It has resulted in increased exposure to cyclophosphamide metabolites in a rabbit model.39 Ranitidine and presumably other H2-receptor antagonists that have little effect on hepatic drug metabolism are not associated with increased cyclophosphamide toxicity.40 Allopurinol increases the half-life of cyclophosphamide and the frequency of leukopenia.41 Cyclophosphamide decreases plasma pseudocholinesterase activity and can potentiate the effect of succinylcholine.42
Chlorambucil
Mechanism of Action
The mechanism of action of chlorambucil is similar to that of cyclophosphamide, but slower.
Pharmacology
Chlorambucil is well absorbed (>70%) after oral administration with peak concentrations occurring within 2 hours.43 Chlorambucil is extensively metabolized by β oxidation to a metabolite, phenylacetic acid mustard, which is also cytotoxic.43,44 Less than 1% of the oral dose of chlorambucil appears in the urine as unchanged drug.41 The plasma half-life of chlorambucil and the phenylacetic acid mustard metabolite is 30 to 180 minutes.43,44
Clinical Indications
Chlorambucil can be effective for patients with inflammatory eye disease including Behçet’s syndrome45,46 and occasionally for refractory dermatomyositis.47 Chlorambucil is used as an alternative alkylating agent for patients unable to tolerate cyclophosphamide because of bladder toxicity or gastrointestinal intolerance. Chlorambucil does not seem to be as effective as cyclophosphamide, however, in the treatment of vasculitis or glomerulonephritis, or systemic sclerosis. The use of chlorambucil in RA is obsolete given the availability of a multitude of alternative drugs with a more favorable risk-benefit profile.
Dosage
Chlorambucil is often started at an oral dose of 0.1 mg/kg/day; the dose is increased or decreased according to clinical response and toxicity. Doses of 0.2 mg/kg/day or greater are associated with more frequent myelosuppression. Alternatively, a “start-low, go-slow” approach, with chlorambucil started at a dose of 4 mg/day and increased in 1-mg increments at 1- to 2-month intervals, if required, may be better tolerated. Even with this approach, 75% of patients discontinued therapy because of chlorambucil-related toxicity.48 Regular monitoring, particularly of the white blood cell count, at approximately 2-week intervals initially, and then monthly when stable, is required.
Toxicity
Hematologic
Myelosuppression is common and may be abrupt in onset. The degree of leukopenia and neutropenia is dose related, but there are considerable interindividual differences in sensitivity. Myelosuppression is usually reversible; however, it may take several months for the white blood cell count to return to the normal range, and some patients remain relatively leukopenic. Irreversible, fatal bone marrow suppression has been reported in patients receiving chlorambucil for rheumatic disease.48
Infection
The average frequency of herpes zoster infection is 13%. As is the case with cyclophosphamide, infections resulting from a wide range of bacterial and nonbacterial pathogens occur.48
Malignancy
Treatment with chlorambucil increases the risk of leukemia, particularly myeloid leukemia, and has been associated with a range of lymphomas.48,49 Various solid organ tumors have occurred in association with chlorambucil, but no causal relationship has been established.48
Purine Analogues
Azathioprine
Structure
Azathioprine is a prodrug that is converted to 6-mercaptopurine involving the removal of an imidazole group.50 6-Mercaptopurine is a purine analogue that acts as a cycle-specific antimetabolite chemotherapeutic agent interfering with the synthesis of nucleotides, thereby inhibiting proliferation of lymphocytes. Azathioprine has a better therapeutic index than 6-mercaptopurine and has replaced it in the treatment of rheumatic autoimmune disease.
Mechanisms of Action
The exact mechanism of action of the active thiopurine metabolites of azathioprine in autoimmune disease is unknown. Thiopurine metabolites such as thioguanine nucleotides decrease the de novo synthesis of purine nucleotides by inhibiting amidotransferase enzymes and purine ribonucleotide interconversion and are incorporated into DNA and ribonucleic acid (RNA).50 The incorporation of thioguanine nucleotides into the nucleic acids of cells is thought to mediate the cytotoxicity of azathioprine, whereas inhibition of purine synthesis may be more important in decreasing cellular proliferation. Leukopenia is unnecessary for immunosuppression. Azathioprine decreases the circulating lymphocyte count, suppresses lymphocyte proliferation, inhibits antibody production, inhibits monocyte production, suppresses natural killer cell activity, and inhibits cell-mediated and humoral immunity.
Pharmacology
Absorption and Distribution
Oral azathioprine is well absorbed and rapidly converted to 6-mercaptopurine, which is further metabolized to several compounds including 6-thiourate (Figure 62-2) that are excreted in urine. The plasma half-life of azathioprine is less than 15 minutes but it is 1 to 3 hours for the active derivative, 6-MP.51 The bioavailability of azathioprine, measured as the concentrations of mercaptopurine achieved after oral administration, varies. In healthy volunteers, bioavailability ranged from 27% to 83% with an average of 47%.51 Mercaptopurine is widely distributed with a volume of distribution of 4 to 8 L/kg.49
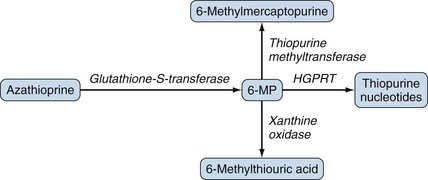
Metabolism and Elimination
The metabolism of azathioprine is complex50,52 and has been simplified in Figure 62-2. Two enzymes, xanthine oxidase and thiopurine methyltransferase (TPMT), shunt mercaptopurine metabolites to relatively inactive compounds, whereas other enzymes such as hypoxanthine-guanine-phosphoribosyl-transferase lead to the formation of cytotoxic thiopurine nucleotides. Low TPMT activity or inhibition of xanthine oxidase by drugs such as allopurinol leads to decreased detoxification and increased formation of cytotoxic metabolites after the administration of azathioprine or mercaptopurine. Maximal concentrations of mercaptopurine occur 1 to 3 hours after administration of azathioprine, and the half-life of mercaptopurine is 1 to 2 hours.51 The half-life of the intracellular, active 6-thioguanine nucleotides is estimated to be 1 to 2 weeks, however, and concentrations do not change over the 24-hour dose period in patients receiving daily azathioprine.53 At conventional rheumatologic doses, approximately 1% of mercaptopurine is excreted unchanged in the urine.53 Increased toxicity can occur with renal impairment (creatinine clearance <25 mL/min), and a modest dose reduction is usually necessary. The substantial interindividual variability in azathioprine disposition and TPMT activity are more important determinants of sensitivity to azathioprine than renal function.54 Azathioprine is only slightly dialyzable (10%) through conventional hemodialysis membranes.
Clinical Indications
In current practice, azathioprine is mainly used in the treatment of connective tissue disease rather than inflammatory joint disease. In RA azathioprine is less effective than methotrexate and has a slow mode of onset when compared with other disease-modifying antirheumatic drugs and biologics. Nevertheless, it remains a treatment option for RA patients with refractory disease or as a corticosteroid-sparing agent in those with organ involvement. Azathioprine is used to treat some patients with lupus nephritis, and although it is more effective than corticosteroids alone, it is not as effective as IV pulse cyclophosphamide to induce remission.55 It is effective in maintenance therapy, however,56 even after low-dose cyclophosphamide induction. For other manifestations of SLE including cutaneous disease, azathioprine is widely used as a glucocorticoid-sparing agent.57 Azathioprine in combination with corticosteroids is useful in the treatment of a range of other autoimmune diseases including inflammatory muscle disease, inflammatory eye disease including Behçet’s syndrome,58 psoriatic arthritis,59 reactive arthritis, and various forms of vasculitis. In systemic vasculitis, azathioprine is less effective as a remission-induction agent than cyclophosphamide15 but safer as a remission-maintenance and glucocorticoid-sparing drug.37 A small study has shown efficacy, however, of high-dose (1200- to 1800-mg infusions) monthly azathioprine as initial treatment of GPA and lupus nephritis.60 Azathioprine is frequently used in systemic sclerosis and overlap syndromes, especially in those with interstitial lung disease or joint involvement.10,61
Toxicity
Hematologic
Reversible myelosuppression is dose related but varies among individuals. Low-dose azathioprine (1 to 2 mg/kg/day) rarely results in leukopenia or thrombocytopenia. Pure red cell aplasia is also rare. Severe myelosuppression is uncommon and caused by low or absent TPMT activity. Decreased TPMT activity leads to a decreased ability to detoxify mercaptopurine and results in increased formation of cytotoxic thioguanine metabolites and clinical toxicity.62 TPMT activity is polymorphic with a trimodal distribution. Approximately 90% of subjects show high activity, 10% show intermediate activity, and 0.3% (the subjects homozygous for the poorly functional polymorphisms) show very low activity.62,63 The median TPMT activity in African-Americans is approximately 17% lower than in white Americans.63 The 1 in 300 subjects with low or absent TPMT activity is at great risk of severe azathioprine-induced myelosuppression, which has a delayed but sudden onset, most commonly 4 to 10 weeks after azathioprine has been started.64 More than half of all cases of leukopenia in patients receiving azathioprine have a normal TPMT genotype and phenotype, however.
Malignancy
Data regarding the risk of malignancy in patients treated with azathioprine for rheumatologic disease are conflicting. Some studies found an increased risk, particularly of lymphoproliferative malignancies, whereas others did not.65 A 24-year retrospective study of 358 SLE patients found no difference in malignancy rates between patients who had received azathioprine and patients who had not, and there were no lymphomas in the azathioprine group.66
Azathioprine Hypersensitivity
Acute hypersensitivity syndromes, usually occurring within 2 weeks of starting therapy, with a range of manifestations including shock, fever, rash, pancreatitis, renal failure, and hepatitis are rare.67
Strategies to Minimize Toxicity
TPMT activity testing is the most commonly used method to identify patients at risk of serious toxicity. More than 23 variants in the TPMT gene, associated with decreased TPMT activity, have been identified, with the TPMT*2, TPMT*3A, and TPMT*3C alleles accounting for most of the intermediate- or low-activity cases. The concordance between TPMT genetics and phenotypes is slightly less than 100%. TPMT activity (phenotype) can be measured directly in red blood cell membranes. Alternatively (e.g., in patients who have undergone blood transfusions), genetic polymorphisms can be identified by polymerase chain reaction. Guidelines for TPMT activity testing vary among different specialties, but it is generally recommended to test TPMT status before starting azathioprine therapy. Whether testing is cost-effective and clinically useful when compared with traditional monitoring of white blood counts is still a matter of debate.68–71 In the absence of TPMT testing, a low initial dose and careful monitoring of the white blood cell count in patients starting azathioprine is required. Some authors suggest weekly monitoring during the first 15 weeks of azathioprine treatment.64 When patients are on a stable dose of azathioprine, blood counts are monitored monthly and liver function tests are monitored every 3 to 4 months.
Drug Interactions
One of the most important, and potentially fatal, drug interactions in rheumatology is the ability of allopurinol, through inhibition of xanthine oxidase–mediated inactivation of mercaptopurine, to increase dramatically the cytotoxic effects of azathioprine and mercaptopurine.72 Various strategies have been employed to treat hyperuricemia and gout in patients receiving azathioprine, a common clinical problem after transplantation. Reduction of the dose of azathioprine by at least two thirds in patients who also are receiving allopurinol is advocated. Because myelosuppression can still occur after a 75% reduction in dose, however, careful monitoring is required.72 Alternatively, uricosurics such as benzbromarone have been effective and safe,72 and MMF has been substituted for azathioprine as an alternative immunosuppressant.73 Combination of azathioprine with several other drugs may also increase the risk of myelosuppression: sulfasalazine, ganciclovir, angiotensin-converting enzyme (ACE) inhibitors, carbamazepine, co-trimoxazole, and clozapine. Azathioprine has been associated with resistance to warfarin in case reports.74
Pregnancy and Lactation
Azathioprine is an FDA Pregnancy Category D drug. Azathioprine and mercaptopurine cross the placenta, but drug and metabolite concentrations are lower in the fetal circulation, suggesting placental metabolism.38,75 There are limited data in rheumatologic diseases, and although it is being used in pregnancy, azathioprine is better avoided in pregnancy and lactation if possible.76 In a prospective observational study in 189 pregnant women treated with azathioprine for various autoimmune conditions, the rate of major malformation was 3.5%, which was similar to the general population.77 Its use was associated with prematurity, however. Azathioprine may be considered in cases where the benefits of disease control in the mother give the best chances of term pregnancy and fetal survival.





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


