118 Hemochromatosis
Hemochromatosis was first recognized in the 1880s in a series of case reports that described “bronze diabetes” and “pigmented cirrhosis,” but von Recklinghausen is credited with the first use of the term in 1889.1 In 1935, the familial pattern of HHC was described by Sheldon,2 who suggested that the disease was due to an inborn error of metabolism. Finch and Finch in 19553 showed that HHC was caused by abnormal iron absorption in the presence of a normal diet. At that time, premorbid recognition of the problem was uncommon, however, and most cases were diagnosed at autopsy. In 1972, serum ferritin became available as a measure of iron stores. Three years later, Simon and colleagues4 discovered that the HHC gene was present on chromosome 6, close to the HLA-A locus. It took 21 more years for the mutated gene, HFE, to be described, and in the last decade, it has been recognized that other gene mutations also can cause iron overload.5 Genetic testing has revolutionized the diagnosis of HHC, although the phenotype of any given mutation may vary greatly.6,7 Nevertheless, the detection of such genes has greatly enhanced the overall prognosis of this condition by allowing the disease to be diagnosed at a preclinical stage in high-risk individuals. This discovery has helped many patients with HHC achieve a normal life expectancy. Improved knowledge at a molecular level has further aided our understanding of the pathogenesis and therapeutic implications of hemochromatosis, permitting many patients with this disease to achieve a normal life expectancy.
Normal Iron Metabolism
The average total body iron content in adults is 3 to 4 g, mostly contained within hemoglobin, but also present in myoglobin and cytochromes, in addition to the storage proteins ferritin and hemosiderin (Table 118-1). Of a typical daily Western diet of 10 to 20 mg of iron, 1 to 2 mg is absorbed by duodenal enterocytes each day.8,9 Heme dietary sources from fish and meat have a higher bioavailability than nonheme sources, such as vegetables. The addition of ascorbic acid to the meal increases absorption of nonheme iron, whereas tannins, bran, and phytates inhibit iron absorption.10,11
Table 118-1 Definitions of Terms Used in Iron Metabolism
| Term | Definitions |
|---|---|
| Ferritin | |
| Transferrin | |
| Transferrin saturation | |
| Iron regulatory proteins | |
| HFE protein | |
| Iron exporter proteins | |
| Hepcidin | Acute phase reactant produced by liver Intrinsic antimicrobial activity Negative regulator of iron absorption Reduces iron release from macrophages Prevents iron loss by reducing entry of iron into bloodstream via inhibition of ferroportin Mutations found in some families with juvenile hereditary hemochromatosis |
| Hemojuvelin | |
| Hemosiderin |
Iron homeostasis is tightly controlled at cellular and molecular levels, influenced by numerous mechanisms, including recent dietary iron intake, the extent of iron stores in the body, and key regulatory peptides. Communication between sites of iron uptake (enterocytes), storage (liver and macrophages), and utilization (erythroid cells) is essential, and an antimicrobial peptide, hepcidin, plays a key role in this regard.12 Hepatic synthesis of hepcidin is stimulated by increases in the body’s iron requirements, as in situations of anemia, hypoxia, or inflammation.13,14 Hepcidin prevents iron loss by reducing the entry of iron into the bloodstream via inhibition of ferroportin, a membrane-bound iron exporter protein found on macrophages, hepatocytes, and enterocytes.14,15 Hepcidin production is downregulated when iron requirements return to normal. In the presence of normal iron stores, iron is retained in the intestinal cells by the protein, mobilferritin, and is subsequently excreted when these cells are shed.
When body iron stores reach an adequate level, ferritin production is increased to facilitate storage, and the transferrin receptor is downregulated to minimize the entry of iron into the cells. The iron responsive element binding protein mediates this process by detaching from ferritin mRNA so that more ferritin can be produced.16 With increasing iron stores, circulating transferrin becomes saturated, and iron is preferentially offloaded to tissue sites that contain cells with high levels of transferrin receptors, such as liver, heart, thyroid, gonads, and pancreatic islet cells.17
Genetics of Hemochromatosis
Four types of HHC have now been described, all linked to gene mutations (Table 118-2).18,19 Classic HHC (type 1) is an autosomal recessive disorder, with a mutation of the HFE gene, located on chromosome 6 (HFE-related HHC). Although numerous such mutations have been described, the most common is a single amino acid substitution of tyrosine for cysteine at position 282 (C282Y). This particular mutation is thought to have arisen in a Celtic/Viking ancestor more than 2000 years ago and is now one of the most common genetic defects in individuals of Northern European origin. This anomaly had no reproductive implications, but may have had survival advantages by protecting against iron deficiency in a susceptible population. Homozygosity for this mutation is a risk factor for organ damage secondary to iron deposition, although phenotypic expression varies widely. Other mutations of the HFE gene include the replacement of histidine with aspartic acid at position 63 (H63D) and the substitution of serine for cysteine at position 65 (S65C). Clinical manifestations of the latter mutations seem to be less serious, although compound heterozygosity of such defects may be associated with evidence of iron overload.
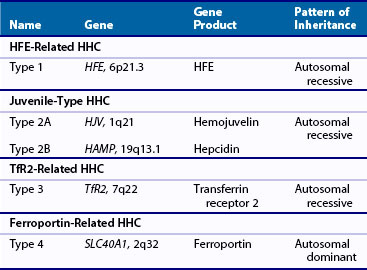
Unlike mutations of the HFE gene that may become clinically obvious in middle age, hemojuvelin (HJV-related HHC, type 1A) or hepcidin (HAMP-related HHC, type 2B) mutations result in juvenile HHC (type 2), which may manifest in the teens or twenties. The rate of iron accumulation seems to be greater than in adult HHC and is often associated with widespread organ involvement and early mortality.20 In contrast to the Northern European inheritance of HFE mutations, juvenile HHC has been most commonly reported in Italy.21 Clinical manifestations of transferrin receptor mutations (TfR2-related HHC, type 3) seem to resemble manifestations of the classic HFE-related HHC. Such mutations are rare, and few cases have been described.22,23
Type 4 or ferroportin-related HHC is an autosomal dominant condition, described in European and Australian families.24,25 Two types of ferroportin mutations have been reported. In the first, loss of surface localization of ferroportin results in a decreased ability of cells to export iron, causing iron to build up predominantly in macrophages. In the second, hepcidin-induced ferroportin dysfunction leads to iron accumulation in parenchymal cells of the liver and other tissues. Phenotypic expression varies, with some patients manifesting the effects of iron overload in a similar manner to classic HHC, and others showing minimal evidence of organ damage.26
Epidemiology
Although HHC previously was thought to be a rare condition, genetic testing has revealed that it is one of the most common heritable disorders. Although 5 out of every 1000 individuals of Northern European origin are homozygous for the HFE mutation, phenotypic expression varies, and clinical cases are much fewer in number. In a study of nearly 100,000 individuals from primary care practices in the United States, the prevalence of C282Y homozygosity was as follows: white, 0.44%; Native American, 0.11%; Hispanic, 0.027%; African-American, 0.014%; Pacific Islander, 0.012%; and Asian, 0.0004%.27 Peak age at the time of diagnosis is 40 to 60 years for classic HHC.
Phenotypic Disease Expression
Clinical manifestations of HHC vary greatly between individuals with similar mutations, suggesting that other factors influence disease expression. One study demonstrated that among C282Y homozygotes, up to 82% have hyperferritinemia, while approximately 28% of male and 1% of females ultimately develop clinical manifestations of HHC—defined as the presence of liver disease, hepatocellular carcinoma, and arthritis of the second and third metacarpophalangeal joints—by the age of 65.28 In contrast, compound heterozygotes with C282Y/H63D mutations exhibit higher serum ferritin and transferrin saturation levels compared with normal controls, but are at very low risk of clinical HHC.29
Other genes may play a role in modifying the phenotypic expression of iron overload. The presence of the gene CYBRD1, which encodes duodenal reductase DCYTB, has been shown to be associated with lower serum ferritin levels in C282Y homozygotes.30 Furthermore, mutations of other iron-related genes, such as hepcidin, hemojuvelin, haptoglobin, and bone morphogenetic protein, may influence disease manifestations.31–35 In addition, profibrotic genes (e.g., TGF) may accelerate the onset of cirrhosis in susceptible individuals.36
Why HHC disease penetrance is more evident in C282Y homozygote males than females may be explained by recurrent menstrual blood loss and consequent slower accumulation of iron stores in women. However, genetically determined sex differences in ferritin levels may occur, as distinct HLA A*03B*07 and A*03B*14 haplotypes have been reported in men and women with clinical evidence of HHC.37
Environmental factors, including diet, smoking, alcohol intake, and comorbid diseases, also influence clinical expression of HHC. The metabolic syndrome is associated with insulin resistance–associated iron overload, which, in the presence of HHC, may have a synergistic effect on liver damage.14 Concurrent liver disease, due to hepatitis or steatosis, may exacerbate the process of fibrogenesis.38 Excess alcohol, meat consumption, and high citrus fruit intake also contribute to increased iron loading. However, ingestion of noncitrus fruits may have a protective effect.39
Pathogenesis
Hepcidin is a key regulatory peptide in the pathogenesis of HHC. Produced by the liver, it acts by binding to ferroportin on enterocytes and macrophages, thereby restricting dietary iron absorption from the gut and inhibiting release of iron recycled by macrophages from aging red cells.12,15 In HHC, inadequate hepcidin synthesis leads to increased intestinal iron absorption and the subsequent deposition of iron in tissues. Absence of hepcidin results in early, severe iron loading, and overexpression of this protein can significantly improve iron deposition in a mouse model of HHC.12,40–42
Chronic iron overload is thought to cause tissue damage via several mechanisms, including weakening of lysosomal membranes and consequent discharge of enzymes into the cytoplasm. Increased free radical formation contributes to lipid peroxidation of cell membranes. The extent and duration of iron deposition correlate with the development of fibrosis, and it is thought that substantial hepatocyte and Kupffer cell iron accumulation precedes organ damage.14 In HHC, iron first accumulates in parenchymal cells, with reticuloendothelial (RE) involvement a late feature, in contrast to transfusional iron overload, in which RE cells are primarily targeted. Values of serum ferritin exceeding 1000 µg/L are associated with significantly increased risks of liver fibrosis and cirrhosis.7,43
Clinical Features
Extra-articular Manifestations
HHC is more common in men than in women and typically manifests in middle-aged adults as iron stores gradually accumulate, often reaching 20 to 30 g. Organ involvement varies and is unpredictable, although the liver, as the major site of iron storage, is typically affected. Commonly, abnormalities of the liver enzymes, checked as part of a routine health screen, are the initial indication of disease. The degree of iron overload has a direct impact on the life expectancy of the affected individual. Without an early diagnosis, progressive fibrosis leading to cirrhosis may occur.44,45 The risk of hepatocellular carcinoma is greatly increased in patients with established cirrhosis.46
Glucose intolerance tends to be a late finding in HHC and is due to progressive iron accumulation in pancreatic beta cells causing low C-peptide and insulin levels. Alpha cell function is usually preserved, however, and serum glucagon levels are normal or increased.47 The risk of diabetes mellitus also is higher in C282Y heterozygotes with no clinical evidence of HHC compared with controls.48
Iron deposition in the heart can result in conduction system abnormalities and heart failure. Several large population studies of HHC and atherosclerosis have failed to find a link. However, elevated ferritin levels, particularly in the setting of nonalcoholic fatty liver disease, may be associated with vascular damage via hepcidin upregulation.49–51
Pituitary involvement in HHC is due to iron deposition, resulting in reduced serum levels of secreted hormones from this gland. Low levels of gonadotropic hormone cause loss of libido and erectile dysfunction.44,52 Hypothyroidism in HHC is thought to be due to a direct toxic effect of iron on thyroid cells and is associated with low thyroxine and elevated thyroid-stimulating hormone.53 Such endocrine abnormalities may contribute to the development of osteoporosis in these individuals.
Patients with HHC have increased susceptibility to certain infections. High serum iron concentrations may increase bacterial virulence, whereas excess iron in macrophages is thought to reduce phagocytosis.54 Particular caution is advised with uncooked seafood because of the risk of septicemia from Vibrio vulnificus. In addition, Yersinia enterocolitica, Listeria monocytogenes, Salmonella enteritidis serotype typhimurium, Klebsiella pneumoniae, Escherichia coli, Rhizopus arrhizus, and Mucor species all have been reported to cause severe illness in patients with iron overload.8
Apart from the increased risk of malignant hepatoma in patients with established cirrhosis is an independent association of nonhepatic cancers, particularly breast and colorectal tumors, in HFE C282Y homozygotes.55 H63D homozygosity confers a threefold increased risk of colorectal cancer in carriers of the MMR gene mutation.56 Iron is potentially carcinogenic via several mechanisms, including its immunosuppressive properties and its role as an essential cofactor for tumor cell growth and in catalyzing the formation of hydroxyl radicals.55 Furthermore, cancer risk is lower with reduced iron stores.57
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


