60 Glucocorticoid Therapy
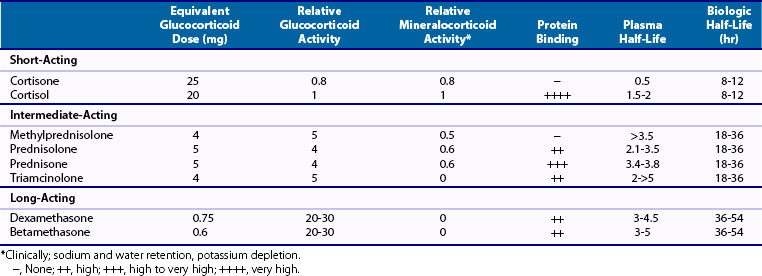
Glucocorticoids differ considerably in potency and biologic half-life.
Glucocorticoids have disease-modifying properties in early rheumatoid arthritis.
The risk of adverse effects of a glucocorticoid is patient, dose, and time dependent.
The risk of adverse effects of low-dose glucocorticoids generally is overestimated.
After local injection of a glucocorticoid, the risk of local bacterial infection is very low.
Low and low-to-moderate doses of prednisolone in pregnancy appear to be safe.
Glucocorticoids are widely used for the treatment of patients with rheumatic disease. The first to be isolated, in 1935, was the naturally occurring glucocorticoid hormone, cortisone. It was synthesized in 1944 and subsequently became available for clinical use. In 1948, cortisone (then called compound E) was administered by the American physician Philip S. Hench to a 29-year-old woman with active rheumatoid arthritis (RA) of longer than 4 years’ duration. Her joints were so painful she could “hardly get out of bed.” After 2 days of treatment with 100 mg of intramuscular compound E daily, “She rolled over in bed with ease, and noted much less muscular soreness.” The next day, she was able to walk with “only a slight limp.” Hench published this case of dramatic improvement in 19491 and won the 1950 Nobel Prize in Physiology or Medicine for his research, which he shared with two colleagues at the Mayo Clinic. Later, by chemical modification of natural steroids, different synthetic glucocorticoids were produced, some of which have proved to be very effective anti-inflammatory and immunosuppressive substances with rapid, sometimes instant, effects.
Characteristics of Glucocorticoids
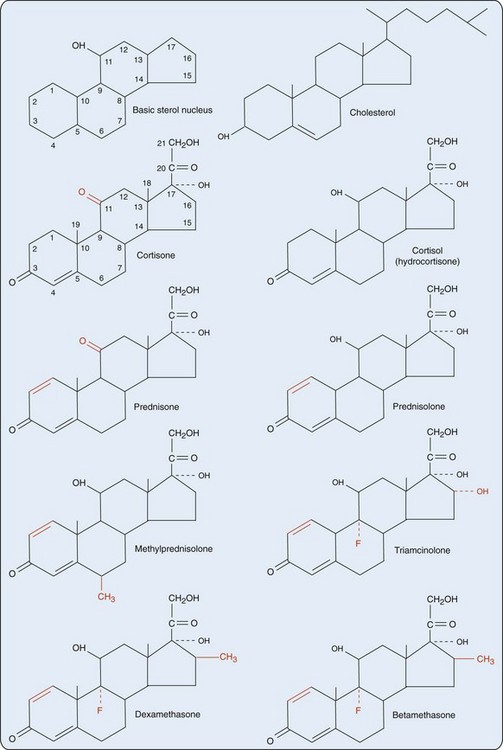
Structure and Classification
The precursor molecule of all steroid hormones is cholesterol, which is also a building block for vitamin D and cell membranes and organelles (Figure 60-1). Steroid hormones and cholesterol are characterized by a sterol skeleton, formed by three six-carbon hexane rings and one five-carbon pentane ring. The carbon atoms of this sterol nucleus are numbered in a specific sequence; the term steroid refers to this basic sterol nucleus (Figure 60-2).
Figure 60-1 Cholesterol as building block for steroid hormones, vitamin D, and cell membranes and organelles.
Steroid hormones can be classified on the basis of their main function into sex hormones (male and female), mineralocorticoids, and glucocorticoids. Sex hormones are synthesized mainly in the gonads, but also in the adrenal cortex. Mineralocorticoids and glucocorticoids are synthesized only in the adrenal cortex; the terms corticosteroid and corticoid for these hormones refer to the adrenal cortex. Some glucocorticoids also have a mineralocorticoid effect and vice versa. The main natural mineralocorticoid is aldosterone, and the main natural glucocorticoid is cortisol (hydrocortisone). Although separation of corticoids into the classes mineralocorticoids and glucocorticoids is not absolute (see later), it is better (more precise) to use the term glucocorticoid than the term corticosteroid when referring to one of these compounds.2 The importance of standardized nomenclature is illustrated by the fact that an electronic literature search can be complicated by multiple synonyms.
In the 1950s, chemical modification of natural steroids revealed numerous structural features essential for specific biologic activities. Synthetic steroid hormones more potent than natural steroid hormones and steroid hormones with altered biologic activity were developed. This research showed that the 17-hydroxy, 21-carbon steroid configuration (see Figure 60-2) is required for glucocorticoid activity through binding to the glucocorticoid receptor. Glucocorticoids with an 11-keto, instead of an 11-hydroxy, group, such as cortisone and prednisone, are prohormones that must be reduced in the liver to their 11-hydroxy configurations. Cortisone is converted by hepatic pathways to cortisol, and prednisone is converted to prednisolone, to become biologically active. Thus in patients with severe liver disease, it is rational to prescribe prednisolone instead of prednisone.
The generation of biologically active glucocorticoids from their inactive forms is promoted by the reductase action of the intracellular enzyme 11β-hydroxysteroid dehydrogenase (11β-HSD) type 1. The same enzyme can by dehydrogenation promote the reverse reaction, leading to inactivation of active glucocorticoids. In contrast, 11β-HSD type 2 has dehydrogenase activity only, so it catalyzes only the conversion of active glucocorticoids to their inactive forms. In different tissues, local balance between the intracellular enzymes 11β-HSD type 1 and type 2 might modulate intracellular glucocorticoid concentrations and thus tissue sensitivity for glucocorticoids.3 Synovial tissue metabolizes glucocorticoids via the two 11β-HSD enzymes, with the net effect being glucocorticoid activation; this increases with inflammation. This endogenous glucocorticoid production in the joint is likely to have an impact on local inflammation and on bone in the joint.4
No qualitative differences have been noted between the glucocorticoid effect of endogenous cortisol and that of exogenously applied synthetic glucocorticoids because these effects are, except for higher doses, predominantly genomic (i.e., mediated through the glucocorticoid receptor).5 However, quantitative differences have been identified. The potency and other biologic characteristics of glucocorticoids depend on structural differences in the steroid configuration. The introduction of a double bond between the 1 and 2 positions of cortisol yields prednisolone, which has about four times more glucocorticoid activity than cortisol (Table 60-1).
Pharmacokinetics and Pharmacology
The affinity of the different glucocorticoids for various plasma proteins varies (see Table 60-1). Of cortisol in plasma, 90% to 95% is bound to plasma proteins, primarily transcortin (also called corticosteroid-binding globulin) and, to a lesser degree, albumin. Protein-bound cortisol is not biologically active, but the remaining 5% to 10% of free cortisol is. Prednisolone has—in contrast to methylprednisolone, dexamethasone, and triamcinolone—a high affinity for transcortin and competes with cortisol for this binding protein. The other synthetic glucocorticoids with little or no affinity for transcortin are two-thirds (weakly) bound to albumin, and about one-third circulate as free glucocorticoid.
Glucocorticoids have biologic half-lives 2 to 36 times longer than their plasma half-lives (see Table 60-1). With a plasma half-life of about 3 hours, prednisolone can be dosed once daily for most diseases. Maximal effects of glucocorticoids lag behind peak serum concentrations. Transcortin binds these compounds more strongly than does albumin. The plasma elimination of glucocorticoids bound to transcortin is slower than that of glucocorticoids that do not bind. Transcortin binding is not a major determinant of biologic half-lives of glucocorticoids, however, in contrast to distribution to different compartments of the body and binding to the cytosolic glucocorticoid receptor. Synthetic glucocorticoids have lower affinity for transcortin but higher affinity for the cytosolic glucocorticoid receptor than does cortisol (see later). The affinity of prednisolone and triamcinolone for the glucocorticoid receptor is approximately two times higher, and for dexamethasone it is seven times higher. Prednisone and cortisone have had negligible glucocorticoid bioactivity before they have been chemically reduced because of their very low affinity for the glucocorticoid receptor.
Another important factor determining biologic half-lives of glucocorticoids is the rate of metabolism. Synthetic glucocorticoids are subject to the same reduction, oxidation, hydroxylation, and conjugation reactions as cortisol. Pharmacologically active glucocorticoids are metabolized primarily in the liver into inactive metabolites and are excreted by the kidneys; only small amounts of unmetabolized drug are also excreted in the urine. An inverse correlation has been noted between prednisolone clearance and age, which means that a given dose may have a greater effect in older individuals.6 Prednisolone clearance also is slower in African-Americans compared with that in whites.7 The serum half-life of prednisolone is 2.5 to 5 hours, but it is increased in patients with renal disease and liver cirrhosis, and in the elderly. Prednisolone can be removed by hemodialysis, but overall, the amount removed does not require dosage adjustment in patients on hemodialysis. In patients with cirrhosis of the liver, clearance of unbound steroid is about two-thirds of normal—a difference that should be taken into account with dosing.
Drug Interactions
Cytochrome P450 (CYP) is a family of isozymes responsible for the biotransformation of several drugs. Drug interactions can be based on induction or on inhibition of these enzymes. Certain drugs (e.g., barbiturates, phenytoin, rifampin) by inducing CYP isoenzymes (e.g., CYP3A4) increase the metabolism (breakdown) of synthetic and natural glucocorticoids, particularly by enhancing hepatic hydroxylase activity, thus reducing glucocorticoid concentrations (Figure 60-3). Rifampin-induced nonresponsiveness to prednisone in inflammatory diseases indeed has been described,8,9 as has rifampin-induced adrenal crisis in patients on glucocorticoid replacement therapy.10 Clinicians should consider increasing the dosage of glucocorticoids in patients who are concomitantly treated with these medications.
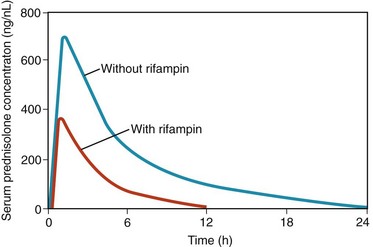
Figure 60-3 Serum prednisolone concentration in time in one patient, after 0.9 mg/kg prednisone orally daily, in the presence and absence of therapy with rifampin. Curve with rifampin, during a period of continuous administration of both drugs. Curve without rifampin, after a washout of rifampin of 4 weeks. Rifampin induces a reduced area under the curve of prednisolone, indicating reduced bioavailability.8
Conversely, concomitant use of glucocorticoids with inhibitors of CYP3A4 (e.g., ketoconazole, itraconazole, diltiazem, mibefradil and grapefruit juice) decreases glucocorticoid clearance and leads to higher concentrations and prolonged biologic half-lives of glucocorticoid drugs, thus increasing the risk of adverse effects.11 Antifungal therapies, especially ketoconazole, on the other hand are known to interfere with endogenous glucocorticoid synthesis and therefore are also used, in doses of 400 to 800 mg per day, to treat hypercortisolism.11 Etomidate, a short-acting intravenous anesthetic agent used for the induction of general anesthesia and for sedation, can also lower cortisol levels, which could be clinically relevant in critically ill patients.11
Concomitant administration of prednisolone and cyclosporine may result in increased plasma concentrations of the former drug; concomitant administration of methylprednisolone and cyclosporine may result in increased plasma concentrations of the latter drug. The mechanism of this probably is competitive inhibition of microsomal liver enzymes. Antibiotics such as erythromycin may increase plasma concentrations of glucocorticoids. Synthetic estrogens in oral contraceptives increase the level of transcortin and thus total (sum of bound and unbound) glucocorticoid levels. Therefore, in women taking oral contraceptives, care is required in the interpretation of cortisol measurements, especially because adrenal insufficiency may be present even when total cortisol levels are within the normal range.12 Next to glucocorticoids, other steroid drugs such as megestrol acetate and medroxyprogesterone inhibit the hypothalamic-pituitary-adrenal axis11; this risk may be increased when they are used concomitantly with glucocorticoids. Sulfasalazine has been reported to increase the sensitivity of immune cells for glucocorticoids,13 which could be beneficial.
Pregnancy and Lactation
If a pregnant woman has to be treated with glucocorticoids, prednisone, prednisolone, and methylprednisolone would be good choices; if the unborn child has to be treated, fluorinated glucocorticoids, such as betamethasone or dexamethasone, would be indicated. Fear of physical (e.g., reduced growth) and neurocognitive adverse effects in children exposed to antenatal repeat doses of 12 mg betamethasone has not been substantiated,14,15 in contrast to postnatal glucocorticoid exposure.16 However, because of a small but increased risk of an oral cleft, it is advised to avoid high doses (1 to 2 mg/kg prednisone equivalent) in the first trimester of pregnancy,17,18 whereas low to moderate doses of prednisone seem to be safe.18
Prednisolone and prednisone are excreted in small quantities in breast milk. Breastfeeding is generally considered safe for an infant whose mother is taking these drugs. Because curves of milk and serum concentrations for prednisolone are virtually parallel in time, exposure of the infant is minimized if breastfeeding is avoided during the first 4 hours after the intake of prednisolone.18
Basic Mechanisms of Glucocorticoids
Genomic and Nongenomic Effects
Glucocorticoids at any therapeutically relevant dosage exhibit pharmacologic effects via classic genomic mechanisms. The lipophilic glucocorticoid passes across the cell membrane, attaches to the cytosolic glucocorticoid receptor and heat shock protein, and binds to glucocorticoid-responsive elements on genomic DNA; it interacts with nuclear transcription factors. This process takes time. When acting through genomic mechanisms, it takes at least 30 minutes before the clinical effect of a glucocorticoid begins to show.19 Only when high doses are given, as in pulse therapy, can glucocorticoids act within minutes by nongenomic mechanisms; this occurs via specific receptor-mediated activity or via nonspecific membrane-associated physicochemical activity.5 The response to high-dose pulse methylprednisolone therapy may be biphasic, consisting of an early, rapid, nongenomic effect and a delayed and more sustained classic genomic effect.20 Clinically, genomic and nongenomic effects cannot be separated, however.
Genomic Mechanisms
Most of the effects of glucocorticoids are exerted via genomic mechanisms by binding to the glucocorticoid receptor located in the cytoplasm of the target cells; glucocorticoids are lipophilic and have a low molecular mass; thus they can pass through the cell membrane easily. Next to the tissue-specific intracellular density of glucocorticoid receptors, the balance of intracellular 11β-HSDs (see earlier) probably determines the sensitivity of specific tissues for glucocorticoids.3 Of the isoforms α and β of the glucocorticoid receptor, only the α isoform, common in all target tissues, binds to glucocorticoids.19 This is a 94-kD protein to which several heat shock proteins (chaperones) are bound. Binding of the glucocorticoid to this complex causes shedding of the chaperones. The resulting activated glucocorticoid receptor–glucocorticoid complex is rapidly translocated into the nucleus, where it binds (as a dimer) to specific consensus sites in the DNA (glucocorticoid-responsive elements), regulating the transcription of a large variety of target genes. This process is termed transactivation. Binding to glucocorticoid-responsive elements results in stimulation or suppression of transcription of these target genes. Suppression of genes also may be mediated by mechanisms involving interaction of the glucocorticoid receptor–glucocorticoid complex (as a momomer) with transcriptional factors, such as activator protein-1 and nuclear factor κB.21 This process is termed transrepression (Figure 60-4).
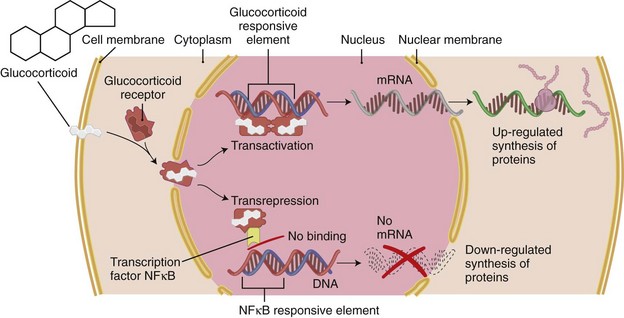
The nature and availability of these transcription factors may be pivotal in determining the differential sensitivity of different tissues to glucocorticoids because they play a crucial role in regulating the expression of a wide variety of proinflammatory genes induced by cytokines. The binding of transcriptional factors to DNA is inhibited by glucocorticoids, resulting in depressed expression of these genes and inhibition of their amplifying role in inflammation. Activated glucocorticoid receptors also may inhibit protein synthesis by decreasing the stability of mRNA through the induction of ribonucleases. This mechanism has been proposed to mediate glucocorticoid-induced inhibition of the synthesis of interleukin (IL)-1, IL-6, granulocyte-macrophage colony-stimulating factor, and inducible cyclooxygenase (COX)-2.22
There is increasing acceptance of the hypothesis that side effects of glucocorticoids, such as diabetes mellitus, osteoporosis, skin atrophy, growth retardation, and cushingoid appearance, may be based predominantly on transactivation of genes after binding of glucocorticoid receptor–glucocorticoid to DNA, whereas the anti-inflammatory effects may be due mostly to the binding of a single glucocorticoid receptor-glucocorticoid complex to transcription factors or co-activators, resulting in gene repression (transrepression). Understanding of these molecular mechanisms may lead to development of novel glucocorticoids, such as selective glucocorticoid receptor agonists, with a more favorable balance of transactivation and transrepression and, clinically, to a more favorable balance of metabolic and endocrine side effects and therapeutic effects21 (see later).
Expression of multiple target genes at the post-transcriptional level, also those influenced by glucocorticoids, is modulated by microRNAs (miRNAs), short noncoding RNA molecules that are implicated in a wide array of cellular and immune processes. Abnormal expression of miRNAs has been found in patients with rheumatoid arthritis. This identifies miRNAs as targets for immunomodulatory drug development.23
Glucocorticoid Effects on the Immune System
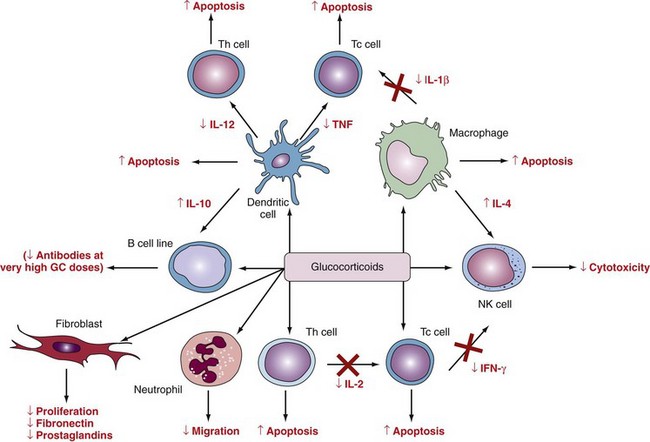
Glucocorticoids reduce activation, proliferation, differentiation, and survival of a variety of inflammatory cells, including macrophages and T lymphocytes, and promote apoptosis, especially in immature and activated T cells (Figure 60-5). This activity is mediated mainly by changes in cytokine production and secretion. In contrast, B lymphocytes and neutrophils are less sensitive to glucocorticoids, and their survival may be increased by glucocorticoid treatment. The main effect of glucocorticoids on neutrophils seems to be inhibition of adhesion to endothelial cells. Glucocorticoids inhibit not only the expression of adhesion molecules, but also the secretion of complement pathway proteins and prostaglandins. At supraphysiologic concentrations, glucocorticoids suppress fibroblast proliferation and IL-1 and tumor necrosis factor (TNF)-induced metalloproteinase synthesis. By these effects, glucocorticoids may retard bone and cartilage destruction in the inflamed joint.24
Leukocytes and Fibroblasts
Administration of glucocorticoids leads to an increase in the total leukocyte count caused by an increase in circulating neutrophil granulocytes in the blood, although the numbers of other leukocyte subsets in blood such as eosinophil and basophil granulocytes, monocytes/macrophages (decreased myelopoiesis and bone marrow release), and T cells (redistribution effect) are decreased. Table 60-2 summarizes the effects of glucocorticoids on leukocyte subsets. The redistribution of lymphocytes, which is maximal 4 to 6 hours after administration of a single high dose of prednisone and returns to normal within 24 hours, has no clinical consequences. B cell function and immunoglobulin production are hardly affected. The effects of glucocorticoids on monocytes and macrophages, including decreased expression of major histocompatibility complex (MHC) class II molecules and Fc receptors, may increase susceptibility to infection, however.25 Effects of glucocorticoids on fibroblasts include decreased proliferation and decreased production of fibronectin and prostaglandins.
Table 60-2 Anti-inflammatory Effects of Glucocorticoids on Immune Cells
| Cell Type | Effects |
|---|---|
| Neutrophils | Increased blood count, decreased trafficking, relatively unaltered functioning |
| Macrophages and monocytes | Decreased blood count, decreased trafficking, decreased phagocytosis and bactericidal effects, inhibited antigen presentation, decreased cytokine and eicosanoid release |
| Lymphocytes | Decreased blood count, decreased trafficking, decreased cytokine production, decreased proliferation and impaired activation, little effect on immunoglobulin synthesis |
| Eosinophils | Decreased blood count, increased apoptosis |
| Basophils | Decreased blood count, decreased release of mediators of inflammation |
Cytokines
The influence of glucocorticoids on cytokine production and action represents one of the major mechanisms of glucocorticoid action in chronic inflammatory diseases such as RA. Glucocorticoids exert potent inhibitory effects on the transcription and action of a large variety of cytokines with pivotal importance in the pathogenesis of RA. Most T helper type 1 (Th1) proinflammatory cytokines are inhibited by glucocorticoids, including IL-1β, IL-2, IL-3, IL-6, TNF, interferon-γ (indicative of Th1 helper cells), IL-17 (indicative of Th17 helper cells), and granulocyte-macrophage colony-stimulating factor (see Figure 60-5). In RA, these cytokines are considered responsible for synovitis, cartilage degradation, and bone erosion. Conversely, the production of Th2 cytokines, such as IL-4, IL-10, and IL-13, may be stimulated or not affected by glucocorticoids (see Figure 60-5).26 These cytokines have been related to the extra-articular features of erosive RA associated with B cell overactivity, such as immune complex formation and vasculitis. Activation of Th2 cells can suppress rheumatoid synovitis and joint destruction through release of the anti-inflammatory cytokines IL-4 and IL-10, which inhibit Th1 activity and downregulate monocyte and macrophage functions.27
Inflammatory Enzymes
An important part of the inflammatory cascade is arachidonic acid metabolism, which leads to the production of prostaglandins and leukotrienes, most of which are strongly proinflammatory. Through the induction of lipocortin (an inhibitor of phospholipase A2), glucocorticoids inhibit the formation of arachidonic acid metabolites. Glucocorticoids also have been shown to inhibit the production of COX-2 and phospholipase A2 induced by cytokines in monocytes/macrophages, fibroblasts, and endothelial cells. In addition, glucocorticoids are potent inhibitors of the production of metalloproteinases in vitro and in vivo, especially collagenase and stromelysin, which are the main effectors of cartilage degradation induced by IL-1 and TNF.28
Adhesion Molecules and Permeability Factors
Pharmacologic doses of glucocorticoids dramatically inhibit exudation of plasma and migration of leukocytes into inflammatory sites. Adhesion molecules play a central role in chronic inflammatory diseases by controlling the trafficking of inflammatory cells into sites of inflammation. Glucocorticoids reduce the expression of adhesion molecules through inhibition of proinflammatory cytokines and by direct inhibitory effects on the expression of adhesion molecules, such as intercellular adhesion molecule-1 and E-selectin.29 Chemotactic cytokines attracting immune cells to the inflammatory site, such as IL-8 and macrophage chemoattractant proteins, also are inhibited by glucocorticoids. Nitric oxide production in inflammatory sites is increased by proinflammatory cytokines, resulting in increased blood flow, exudation, and probably amplification of the inflammatory response. The inducible form of nitric oxide synthase by cytokines is potently inhibited by glucocorticoids.30
Hypothalamic-Pituitary-Adrenal Axis
Pathophysiology
Proinflammatory cytokines, such as IL-1 and IL-6, and eicosanoids, such as prostaglandin E2, and endotoxins all activate corticotropin-releasing hormone (CRH) at the hypothalamic level (Figure 60-6). This activation stimulates the secretion of adrenocorticotropic hormone (ACTH) by the pituitary gland and of glucocorticoids by the adrenal glands. In otherwise healthy individuals with severe infection or other major physical stress, cortisol production may increase to six times the normal amount.12 In patients with active RA (or other chronic inflammatory diseases), the increase in cortisol driven by elevated cytokines might be inappropriately low,31 meaning that cortisol levels—although normal or elevated in the absolute sense—are insufficient to control the inflammatory response. This is the concept of relative adrenal insufficiency.31–33 Endogenous and exogenous glucocorticoids exert negative feedback control on the hypothalamic-pituitary-adrenal axis directly by suppressing secretion of ACTH and CRH, and indirectly by suppressing release from inflammatory tissues of proinflammatory cytokines, which stimulate secretion of ACTH and CRH (see Figure 60-6). Sensitivity of the hypothalamic-pituitary-adrenal axis for proinflammatory cytokines is probably decreased in RA.34
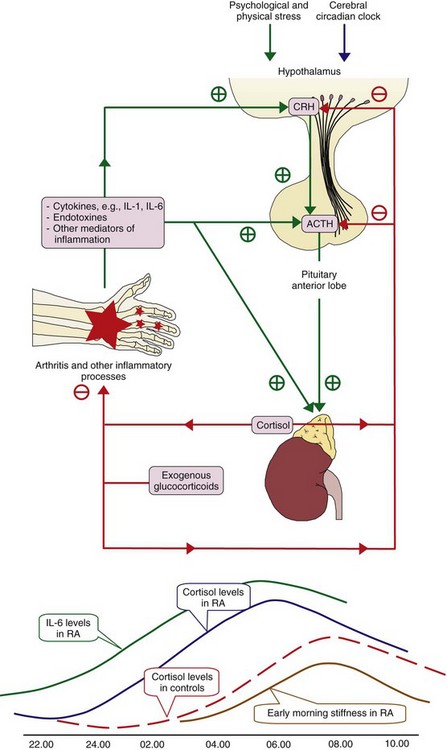
ACTH is secreted in brief, episodic bursts, resulting in sharp increases in plasma concentrations of ACTH and cortisol, followed by slower declines in cortisol levels—the normal diurnal rhythm in cortisol secretion. Secretory ACTH episodic bursts increase in amplitude but not in frequency after 3 to 5 hours of sleep, reach a maximum during the hours before and the hour after awakening, decline throughout the morning, and are minimal in the evening. Cortisol levels are highest at about the time of awakening in the morning, are low in the late afternoon and evening, and reach their lowest level some hours after falling asleep (see Figure 60-6). Glucocorticoids are not stored in the adrenal glands in significant quantities. Continuing synthesis and release are required to maintain basal secretion or to increase blood levels during stress. The total daily basal or physiologic secretion of cortisol in humans has been estimated to range from 5.7 to 10 mg/m2/day.35,36 This would be covered in primary adrenal insufficiency by oral administration of 15 to 25 mg cortisol,35 equivalent to about 4 to 6 mg prednisone. This low daily cortisol production rate may explain the cushingoid symptoms and other adverse effects that are sometimes observed in patients with adrenal insufficiency who are using glucocorticoids at doses previously regarded to be replacement doses (based on estimates of physiologic secretion of cortisol of 12 to 15 mg/m2/day), which are in fact supraphysiologic doses.
Effects of Glucocorticoids on the Hypothalamic-Pituitary-Adrenal Axis
Chronic suppression of the hypothalamic-pituitary-adrenal axis by administration of exogenous glucocorticoids leads by negative feedback loops on CRH and ACTH (see Figure 60-6) to failure in pituitary ACTH release, and thus to partial functional adrenal atrophy with loss of cortisol secretory capability in the fasciculata-reticularis zone. This inner cortical zone is the site of cortisol and adrenal androgen synthesis and is dependent on ACTH for structure and function. The outer cortical (glomerulosa) zone is involved in mineralocorticoid (aldosterone) biosynthesis and is functionally independent of ACTH. It stays functionally intact. Patients have failure of pituitary ACTH release and adrenal responsiveness to ACTH. Serum cortisol, ACTH levels, and adrenal responsiveness to ACTH are low, but other pituitary axes function normally, in contrast to the situation in most primary pituitary disorders. The time required to achieve suppression depends on the dosage and the serum half-life of the glucocorticoid used, but it also varies among patients, probably because of individual differences in glucocorticoid sensitivity and rates of glucocorticoid metabolism. Prediction with certainty of chronic suppression of the hypothalamic-pituitary-adrenal axis and adrenal insufficiency is impossible. This risk may be increased when glucocorticoids are used concomitantly with other steroid drugs such as megestrol acetate and medroxyprogesterone, inhibiting the hypothalamic-pituitary-adrenal axis.11
The duration of the anti-inflammatory effect of one dose of a glucocorticoid approximates the duration of hypothalamic-pituitary-adrenal suppression. After a single oral dose of 250 mg of hydrocortisone or cortisone, 50 mg of prednisone or prednisolone, or 40 mg of methylprednisolone, suppression for 1.25 to 1.5 days has been described. Duration of suppression after 40 mg of triamcinolone and 5 mg of dexamethasone was 2.25 and 2.75 days.37 After intramuscular administration of a single dose of 40 to 80 mg of triamcinolone acetonide, the duration of hypothalamic-pituitary-adrenal suppression is 2 to 4 weeks, and after 40 to 80 mg of methylprednisolone, suppression lasts 4 to 8 days.37
In the case of long-term therapy, for patients who have had less than 10 mg of prednisone or its equivalent per day in one dose in the morning, the risk of clinical (symptomatic) adrenal insufficiency is not high, but neither is it negligible. A review of adrenal insufficiency stated that if the daily dose is 7.5 mg of prednisolone or equivalent or more for at least 3 weeks, adrenal hypofunction should be anticipated, and acute cessation of glucocorticoid in this situation could lead to problems.12 Patients who have received glucocorticoids for less than 3 weeks or have been treated with alternate-day prednisolone therapy do not have zero risk of suppression of the hypothalamic-pituitary-adrenal axis, depending on the dose,38,39 but the risk is low. After 5 to 30 days of at least 25 mg of prednisone or equivalent daily, suppression of adrenal response (measured by a low-dose corticotropin test) was present in 34 of 75 patients studied (45%).40 In these patients, a basal plasma cortisol concentration less than 100 nmol/L was highly suggestive of adrenal suppression, whereas levels of basal cortisol greater than 220 nmol/L predicted a normal adrenal response in most, but not all, patients. When in doubt, it seems prudent to treat patients as having secondary adrenal insufficiency. Secondary adrenal insufficiency generally has a less dramatic presentation than primary adrenal insufficiency because aldosterone levels, which are controlled predominantly by the renin-angiotensin system, are preserved; mineralocorticoid therapy is not necessary.
Treatment with Glucocorticoids
Glucocorticoids are widely used in various dosages for several rheumatic diseases. Often it is unclear what is meant by the semi-quantitative terms used for dosages, such as low or high. Based on pathophysiologic and pharmacokinetic data, standardization has been proposed to minimize problems in interpretation of these generally used terms (Table 60-3).2
Table 60-3 Terminology of Dosages of Glucocorticoids for Use in Rheumatology
| Low dose | ≤7.5 mg prednisone or equivalent per day |
| Medium dose | >7.5 mg, but ≤30 mg prednisone or equivalent per day |
| High dose | >30 mg, but ≤100 mg prednisone or equivalent per day |
| Very high dose | >100 mg prednisone or equivalent per day |
| Pulse therapy | ≥250 mg prednisone or equivalent per day for 1 day or a few days |
Indications
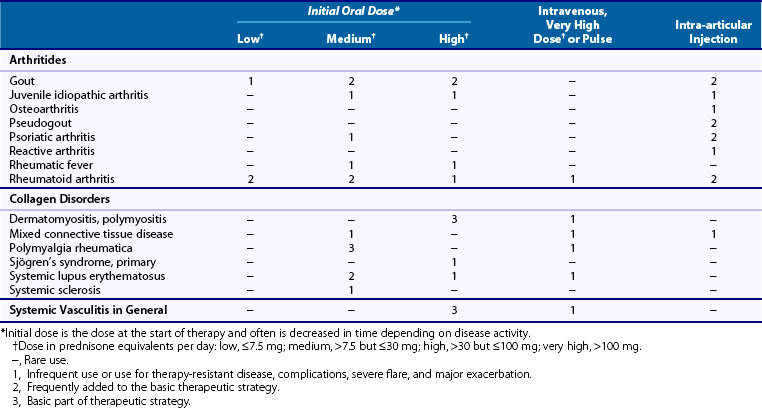
For each disease, indications for glucocorticoid therapy are discussed in the specific chapters. An overview is given here (Table 60-4), which summarizes only the general uses and dosages of glucocorticoids. Without detailed description, some of the indications could be considered questionable at first glance. In systemic sclerosis, glucocorticoids, especially in high doses, are contraindicated because of the risk of scleroderma renal crisis, but they may be useful for myositis or interstitial lung disease. Glucocorticoids are a basic part of the therapeutic strategy in myositis, polymyalgia rheumatica, and systemic vasculitis. For other diseases, glucocorticoids serve as adjunctive therapy or are not used at all. For instance in RA, glucocorticoids are almost exclusively used as adjunctive therapy in combination with other disease-modifying antirheumatic drugs (DMARDs) (see later). In osteoarthritis, glucocorticoids are not given except for intra-articular injection if signs of synovitis of the osteoarthritic joint are present.41 For generalized soft tissue disorders, glucocorticoids are not indicated, and for localized soft tissue disorders, they should be used only for intralesional injection.42
Glucocorticoid Therapy in Rheumatoid Arthritis
Glucocorticoids are a frequently applied medication in RA. In the past, more patients with RA seemed to be given concomitant glucocorticoids in the United States than in Europe—54% versus 27%43,44—whereas more recent data suggest that 38% of RA patients in the United States use glucocorticoids45 versus up to 55% of German RA patients.46 Aims of this therapy include reduction of signs and symptoms and inhibition of joint damage.
Signs and Symptoms
As can be seen in Table 60-4, RA is the only disease in which glucocorticoid therapy is often started and maintained at a low dose as additional therapy. The rationale for this therapy is a probable, relative insufficiency of the adrenal gland in patients with active RA.31 Glucocorticoids are highly effective for relieving symptoms in patients with active RA in doses of less than 10 mg/day. Many patients become functionally dependent on this therapy, however, and continue it over the long term.47 A review of seven studies (253 patients) concluded that glucocorticoids, when administered for approximately 6 months, are effective for the treatment of RA.48 After 6 months of therapy, the beneficial effects of glucocorticoids seem to diminish. If this therapy then is tapered off and stopped, however, patients often—over some months—experience aggravation of symptoms.
Radiologic Joint Damage: Glucocorticoids as DMARDs
In 1995, joint-preserving effects of 7.5 mg of prednisolone daily for 2 years were described in patients with RA of short and intermediate duration who also were treated with DMARDs. The group of RA patients participating in this randomized, placebo-controlled trial was heterogeneous, not only with respect to disease duration, but also with respect to stages of the disease and types and dosages of DMARDs.49 In another trial published in 1997, patients with early RA were randomly assigned to step-down therapy with two DMARDs (sulfasalazine and methotrexate) and prednisolone (start 60 mg/day, tapered in six weekly steps to 7.5 mg/day and stopped at 34 weeks) or to sulfasalazine alone. In the combined drug strategy group, a statistically significant and clinically relevant effect in retarding joint damage was shown compared with the effect of sulfasalazine alone.50 In an extension of this study, long-term (4 to 5 years) beneficial benefits were shown regarding radiologic damage after the combination strategy.51 It has been hypothesized that the superior effect of the combination therapy in this trial can be ascribed to prednisolone because in three double-blind, randomized trials, the effect of the combination of methotrexate and sulfasalazine was not superior to that of either drug alone.52–54
In a German study, 200 patients with early RA were treated with methotrexate or intramuscular gold and were randomly assigned to additional treatment with 5 mg of prednisolone or placebo. After 2 years, progression of radiologic damage proved to be less in the prednisolone-treated patients than in those treated with placebo.55 In 2002, results of the Utrecht study on the effects of prednisolone in DMARD-naïve patients with early RA were published. This is the only placebo-controlled trial in which prednisolone was applied as monotherapy as the first step. The progression of radiologic joint damage was inhibited by 10 mg of prednisolone daily in these patients (who received DMARD therapy only as rescue).56
The Utrecht study reported a 40% decreased need for intra-articular glucocorticoid injections, a 49% decreased need for acetaminophen use, and a 55% decreased need for nonsteroidal anti-inflammatory drugs (NSAIDs) in the prednisolone group compared with the placebo group. This indicates that in clinical trials evaluating the clinical effects of DMARDs or glucocorticoids, additional therapies should be taken into account. In an extension of this study, at 3 years after the end of the study and 2 years after tapering off and stopping the prednisolone therapy, beneficial radiologic benefits of prednisolone were still present (Figure 60-7).57
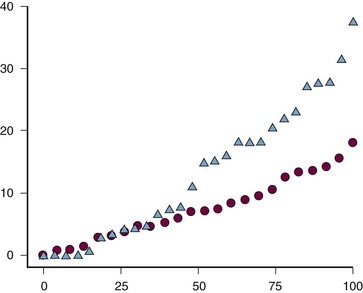
Figure 60-7 Cumulative probability plot of mean yearly radiographic progression over 3 years since the end of the original 2-year study in patients originally randomized to receive prednisone therapy (triangles) or placebo (circles).56,57 At the end of the 2-year trial, the prednisone therapy was tapered down and stopped, if possible. Y-axis, Yearly progression of radiographic joint damage according to the van der Heijde modification of the Sharp method.
In another 2-year study in 250 patients with early RA, 7.5 mg/day of prednisolone added to DMARD therapy retarded joint damage and increased the remission rate compared with placebo added to DMARDs.58 Even in an intensive treat-to-target methotrexate-based strategy in early RA, prednisone enhanced clinical efficiency and reduced erosive joint damage.58a
Negative studies on the effects of glucocorticoids on radiologic damage have also been published,59–61 but in early RA, evidence of glucocorticoid joint-sparing effects, which persist after therapy is stopped, seems convincing, thus classifying glucocorticoids as DMARDs. A meta-analysis on radiographic outcome analyzed 15 studies (two with negative results) that included a total of 1414 patients. Because different methods had been used in the individual trials, radiographic scores were expressed as a percentage of the maximum possible score for the specific radiographic method used. The standardized mean difference in progression was 0.40 in favor of strategies using glucocorticoids (95% confidence interval, 0.27 to 0.54). This was considered a conservative estimate because the most conservative estimate of the difference in each study had been chosen.62
It is still unknown, however, whether glucocorticoids can also inhibit progression of erosion in RA of longer duration than 2 years. A so-called window of opportunity may exist in the treatment of RA.63 If this window is present, effective treatment of early RA with glucocorticoids and DMARDs may result in an effect that lasts for a long time and in disease that is easier to control, whereas if effective treatment starts later, this opportunity may be lost, resulting in more difficult control of the disease with inflammation fueled by joint damage. Most studies on glucocorticoids and radiologic damage employed a dose of 5 to 10 mg/day of prednisone equivalent during 2 years, but a scheme starting with 60 mg/day tapered off and stopped within 34 weeks also was effective. In addition, because glucocorticoid-induced osteoporosis and peptic ulcer complications (if glucocorticoids are combined with nonsteroidal anti-inflammatory drugs [NSAIDs]) can be prevented much more effectively now than some decades ago, the joint-protective effect of prednisolone in RA during the first 2 years of the disease in a dose of 5 to 10 mg daily is a relevant finding.
The joint-sparing effect of glucocorticoids probably is based on inhibition of proinflammatory cytokines such as IL-1 and TNF,64 which stimulate osteoblasts and T cells to produce receptor activator of nuclear factor κB (RANK) ligand. This binds to RANK on osteoclast precursor cells and on mature osteoblasts, leading to activation of osteoclasts, which are responsible for bone resorption, periarticular osteopenia, and formation of bone erosions in RA.
A toxicity index score for DMARDs was published (based on symptoms, laboratory abnormalities, and hospitalization data) after evaluation of 3000 patients with more than 7300 patient-years from the Arthritis, Rheumatism, and Aging Medical Information System (ARAMIS) database.65 Although this score has not been validated and is influenced by confounding-by-indication, it gives an impression of the relative toxicity of glucocorticoids. It is comparable with that of other immunosuppressive medications used in RA, such as methotrexate and azathioprine. A review also showed that the incidence, severity, and impact of adverse effects of low-dose glucocorticoid therapy in RA trials were modest and suggested that probably many of the well-known adverse effects of glucocorticoids are predominantly associated with high-dose treatment.66
Because many questions remain to be answered, such as how the effects of glucocorticoids compare with those of high dosages of methotrexate or of TNF blockers, and for how long glucocorticoids should be prescribed and in what dosages, the final place of glucocorticoid therapy in RA has to be clearly determined. Nevertheless in early RA the use of glucocorticoids generally has been accepted.66a Guidelines on how to use (low-dose) glucocorticoids and how to monitor this therapy have been developed.67,68
Prevention of Early (Rheumatoid) Arthritis Development with Glucocorticoids
Recently, trials have been done to try to prevent arthralgia or early arthritis from progressing to chronic arthritis. In patients with (very) early arthritis or individuals with arthralgia and antibodies to citrullinated proteins or rheumatoid factor, intramuscular glucocorticoid injections did not prevent arthritis development in two placebo-controlled trials,69,70 but in another placebo-controlled double-blind trial these injections postponed the need for DMARDs and prevented 1 in 10 patients from progressing into RA at assessment at 12 months.71 These results are preliminary and nonconclusive, and it is clear that further resarch is needed.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


