88 Giant Cell Arteritis, Polymyalgia Rheumatica, and Takayasu’s Arteritis
Giant cell arteritis affects adults older than 50 years.
The diagnosis of giant cell arteritis is usually confirmed by temporal artery biopsy.
Early treatment of giant cell arteritis can prevent blindness.
Polymyalgia rheumatica can occur by itself or with giant cell arteritis.
Takayasu’s arteritis most frequently affects the aorta and its major branches in young women.
Giant Cell Arteritis and Polymyalgia Rheumatica
American College of Rheumatology Criteria
Classification criteria for the diagnosis of GCA have been proposed by the American College of Rheumatology (ACR) (Table 88-1).1 Three classification schemes have been proposed for PMR (Table 88-2).2,3,3a The provisional criteria for 2012 were developed in part to incorporate information from ultrasound evaluation.
Table 88-1 American College of Rheumatology Classification Criteria for Giant Cell Arteritis
| Criterion* | Definition |
|---|---|
| Age at disease onset ≥ 50 yr | Development of symptoms or findings beginning at age 50 or older |
| New headache | New onset or new type of localized pain in the head |
| Temporal artery abnormality | Temporal artery tenderness to palpation or decreased pulsation, unrelated to arteriosclerosis of cervical arteries |
| Elevated ESR | ESR ≥50 mm/hr by the Westergren method |
| Abnormal artery biopsy | Biopsy specimen with artery showing vasculitis characterized by a predominance of mononuclear cell infiltration or granulomatous inflammation, usually with multinucleated giant cells |
ESR, erythrocyte sedimentation rate.
* For purposes of classification, a patient with vasculitis is said to have giant cell (temporal) arteritis if at least three of these five criteria are present. The presence of any three or more criteria yields a sensitivity of 93.5% and a specificity of 91.2%.
From Hunder GG, Bloch DA, Michel BA, et al: The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis, Arthritis Rheum 33:1125, 1990.
Table 88-2 Diagnostic* and Classification Criteria for Polymyalgia Rheumatica
| Diagnostic Criteria of Chuang and Colleagues2 (1982) |
| Diagnostic Criteria of Healey3 (1984) |
| Classification Criteria of Dasgupta and Colleagues3a (2012) |
| Age 50 years or older, bilateral shoulder aching and abnormal C-reactive protein and/or ESR† |
| Points without US (0-6) | Points with US‡ (0-8) | |
|---|---|---|
| Morning stiffness duration >45 min | 2 | 2 |
| Hip pain or limited range of motion | 1 | 1 |
| Absence of rheumatoid factor or anticitrullinated protein antibody | 2 | 2 |
| Absence of other joint involvement | 1 | 1 |
| At least one shoulder with subdeltoid bursitis and/or biceps tenosynovitis and/or glenohumeral synovitis (either posterior or axillary) and at least one hip with synovitis and/or trochanteric bursitis | Not applicable | 1 |
| Both shoulders with subdeltoid bursitis, biceps tenosynovitis or glenohumeral synovitis | Not applicable | 1 |
ESR, erythrocyte sedimentation rate; US, ultrasound.
* For each set of criteria, all the findings must be present for polymyalgia rheumatica to be diagnosed.
† A score of 4 or more is categorized as polymyalgia rheumatica in the algorithm without US, and a score of 5 or more is categorized as polymyalgia rheumatica in the algorithm with US.
‡ Optional ultrasound criteria.
From Salvarani C, Cantini F, Boiardi L, Hunder GG: Polymyalgia rheumatic and giant-cell arteritis, N Engl J Med 347:261, 2002; and Dasgupta B, Cimmino MA, Kremers HM, et al: 2012 provisional classification criteria for polymyalgia rheumatica: a European League Against Rheumatism/American College of Rheumatology collaborative initiative, Arthritis Rheum 64:943–954, 2012.
Definitions
Giant Cell Arteritis
GCA is the most common form of systemic vasculitis in adults.4 The disease affects primarily the extracranial branches of the carotid artery in patients older than 50 years. The most feared complication of GCA is irreversible loss of vision. Because the cause of GCA is unknown, various names—including temporal arteritis, cranial arteritis, and granulomatous arteritis—have been used to highlight different salient features.5 All the names for this disease have both merits and shortcomings. The designation temporal arteritis or cranial arteritis, for example, conveys how frequently the temporal arteries or other cranial arteries are involved but fails to capture GCA’s more widespread nature. Although granulomatous arteritis and GCA pay homage to an important pathologic finding, this focus is undeserved because giant cells are absent in about half the cases and may be present in other forms of vasculitis. With no perfect name available, this chapter bows to convention and refers to this disease as GCA.
Polymyalgia Rheumatica
Polymyalgia rheumatica, a term suggested by Barber, is a syndrome characterized by aching in the proximal portions of the extremities and torso. Because there are no specific diagnostic tests or pathologic findings, PMR is defined by its clinical features. The features included in most definitions of PMR are as follows (1) aching and morning stiffness lasting half an hour or longer in the shoulder, hip girdle, neck, or some combination; (2) duration of these symptoms for 1 month or longer; (3) age older than 50 years; and (4) laboratory evidence of systemic inflammation such as an elevated erythrocyte sedimentation rate (ESR).2 Some definitions also include a rapid response to small doses of glucocorticoids such as prednisone 10 mg/day.6 The presence of another specific disease other than GCA such as rheumatoid arthritis (RA), chronic infection, polymyositis, or malignancy excludes the diagnosis of PMR.
Epidemiology
The incidence of GCA varies widely in different populations, from less than 0.1 per 100,000 to 77 per 100,000 persons aged 50 years and older.4,7–11 The greatest risk factor for developing GCA is aging; the disease almost never occurs before age 50, and its incidence rises steadily thereafter. The average age at diagnosis has risen over the past half century, from 74.7 years in the 1950s to 79.2 years now.12 Nationality, geography, and race are also important, with the highest incidence figures found in Scandinavians and in Americans of Scandinavian descent. The lowest incidence of GCA is reported in Japanese, northern Indians, and African-Americans. In western Europe, GCA is more common in the northern latitudes than the southern ones. The incidence of GCA has been increasing over the past 20 to 40 years, possibly because of greater physician awareness.4,7 Some studies have reported seasonal variations and clustering of cases, with peaks about 7 years apart.7,10 The prevalence of GCA in Olmsted County, Minnesota, home to many Scandinavian immigrants, is 200 per 100,000 population aged 50 years or older. Autopsy studies suggest that GCA may be more common than is clinically apparent. Östberg13 found arteritis in 1.6% of 889 postmortem cases in which sections of the temporal artery and two transverse sections of the aorta were examined.
Genetic susceptibility to the development of GCA was initially suggested by reports of GCA in families14 and, more recently, by studies demonstrating an association with genes in the human leukocyte antigen (HLA) class II region.15 Sixty percent of GCA patients have HLA-DRB1*04 haplotype variants, which have a common sequence motif in the second hypervariable region of the B1 molecule.15 This motif differs from that found in patients with RA.15 The low prevalence of these alleles in African-Americans may explain why blacks develop GCA relatively infrequently. To date, GCA is the form of systemic vasculitis most closely associated with HLA class II genes. Susceptibility to GCA and PMR has also been associated with polymorphisms of genes for tumor necrosis factor, intercellular adhesion molecules, and interleukin 18 (IL-18).4
The existence of environmental risk factors has been suggested by the geographic clustering of GCA cases. Smoking appears to increase the risk of developing GCA sixfold in women.16 Circumstantial evidence links the development of GCA to a variety of infectious agents including Mycoplasma pneumoniae, varicella-zoster virus, parvovirus B19, and parainfluenza virus type I.10,11 The results of examining temporal artery biopsy specimens with polymerase chain reaction methods to detect parvovirus B19 or herpesvirus DNA have been negative or inconsistent.17 The reported association between GCA and Chlamydia pneumoniae infection has not withstood close scrutiny.18
Gender and health status also influence the development of GCA. Women are affected about twice as often as men.10 Having diabetes reduces the risk of developing GCA by 50% in women.2 Although patients with GCA have an increased risk of developing thoracic aortic aneurysms, they do not have overall higher mortality rates.10
PMR is two to three times more common than GCA.4,8,9,19 In Olmsted County, Minnesota, 245 cases of PMR were diagnosed during the 22-year period from 1970 through 1991, providing an average annual incidence rate of 52.5 cases per 100,000 persons aged 50 years or older.19 The prevalence of PMR (active plus remitted cases) was approximately 600 per 100,000 persons aged 50 years and older.19 PMR is associated with the same HLA-DR4 genes as GCA.15,20
Cause, Pathology, and Pathogenesis
In GCA, inflammation is found most often in medium-size muscular arteries that originate from the arch of the aorta.10,13,21,22 The inflammation tends to affect the arteries in a segmental fashion (possibly leading to “skip lesions” within arteries), but long portions of arteries may be involved.23 In patients who died during the active phase of GCA, the greatest frequency of severe involvement was noted in the superficial temporal arteries, vertebral arteries, and ophthalmic and posterior ciliary arteries.24 The internal carotid, external carotid, and central retinal arteries were affected somewhat less frequently.24 In other postmortem studies, lesions were commonly found in the proximal and distal aorta and internal and external carotid, subclavian, brachial, and abdominal arteries.13 Because GCA affects vessels with an internal elastic lamina and vasa vasorum, and because intracranial arteries lose these structures after penetrating the dura, it is not surprising that GCA rarely involves intracranial arteries.24–26 In some patients with GCA, follow-up biopsy or autopsy surveys showed the persistence of mild chronic inflammation, even though symptoms had resolved.2
Early in the disease, collections of lymphocytes are confined to the region of the internal or external elastic lamina or adventitia. The inflammation may be limited to the vasa vasorum in some cases.3 Intimal thickening with prominent cellular infiltration is a hallmark of more advanced cases. In heavily involved areas, all layers are affected (Figure 88-1). Transmural inflammation of portions of the arterial wall (including the elastic laminae) and granulomas containing multinucleated histiocytic and foreign body giant cells, histiocytes, lymphocytes (which are predominantly CD4+ T cells), and some plasma cells and fibroblasts are found.22,27,28 Eosinophils may be seen, but polymorphonuclear leukocytes are rare. Thrombosis may develop at sites of active inflammation; later, these areas may recanalize. The inflammatory process is usually most marked in the inner portion of the media adjacent to the internal elastic lamina. Fragmentation and disintegration of elastic fibers occur, closely associated with an accumulation of giant cells (Figure 88-2). However, giant cells are seen in only about half of routinely examined specimens; therefore they are not required to make the diagnosis if other features are compatible. In contrast to some other forms of systemic vasculitis (e.g., polyarteritis nodosa, microscopic polyangiitis, granulomatosis with polyangiitis [GPA] [formerly Wegener’s granulomatosis]), fibrinoid necrosis is rarely if ever observed in GCA.3
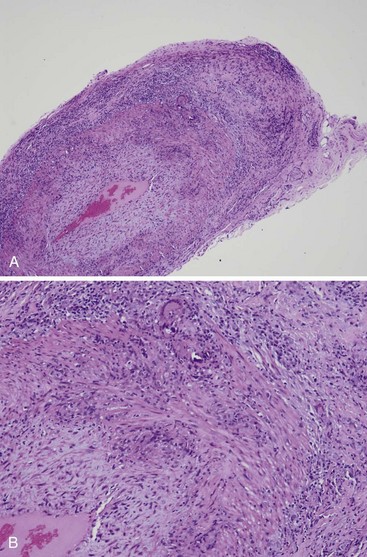

Immunohistochemical studies demonstrate inflammatory changes that are specific for each layer of the affected artery.11,22,28–31 Dendritic cells, which can present antigen and activate T cells, are found in the adventitia, along with two separate lineages of CD4+ T cells: (1) Th1 cells that secrete interferon-γ (IFN-γ) and interleukin-2 (IL-2) and (2) Th17 cells that secrete interleukin-17 (IL-17) family members including IL-17A.32–34 The adventitial T cells show evidence of clonal expansion. The adventitia is also infiltrated with macrophages that secrete IL-1, IL-6, and transforming growth factor-β (TGF-β). The media is populated mostly by macrophages that, in contrast to those in other layers, produce matrix metalloproteinases and oxygen free radicals. Closer to the intima, the macrophages secrete nitric oxide and unite to form syncytia—the giant cells—which produce platelet-derived growth factor (PDGF) and substances that stimulate intimal proliferation.22,35
Although microscopic examination of arteries in PMR is usually normal, immunohistochemical studies of apparently uninvolved temporal arteries reveal upregulation of the same macrophage-related inflammatory cytokines found in GCA.22 The T cell cytokine IFN-γ is abundantly expressed in GCA and is absent in arteries from patients having only PMR. Pathologically, relatively little else has been found in the arteries of patients with PMR. Granulomatous myocarditis and hepatitis have been noted.36 Muscle biopsy specimens may be normal or show nonspecific type II muscle atrophy. However, a number of reports have shown the presence of lymphocytic synovitis in the knees, sternoclavicular joints, and shoulders and evidence of a similar reaction in sacroiliac joints.37,38 Synovitis (mostly subclinical) was shown in bone scans demonstrating an increased uptake of technetium pertechnetate in the joints of 2 of 25 patients with PMR.37 More sensitive studies using magnetic resonance imaging (MRI) and ultrasonography have convincingly demonstrated that in PMR, the principal foci of inflammation are the bursae surrounding the shoulder more than the glenohumeral joint itself.5 Sera from patients with GCA, PMR, or both demonstrate evidence of systemic inflammation, with increased levels of circulating immune complexes during active disease39 and elevated levels of IL-6 and IL-1.7
These observations, together with the results from experiments in which temporal arteries from patients with GCA have been implanted into mice with severe combined immunodeficiency (SCID), suggest that GCA results from an adaptive immune response in the wall of an artery.22,33 In this model (Figure 88-3) the key initiating event is activation of dendritic cells located in the adventitia, the only arterial layer normally penetrated by the vasa vasorum. In large and medium-size arteries, immunohistochemical studies reveal that the dendritic cells in temporal arteries have a specific phenotype and express fascin and CD11c.22,32 Dendritic cells express Toll-like receptors (TLRs) that normally serve as sentinels, monitoring for any immunologic breach of the adventitia.40 In GCA, activation of TLRs on dendritic cells appears to be the initial triggering event.32,40,41 Of the many different types of TLRs, TLR-2 and TLR-4 might be the most important in GCA. Although the exact trigger of the adaptive immune response in GCA is not known, experiments in the GCA-SCID mouse model have shown that blood-borne lipopolysaccharide serves as an effective TLR ligand in temporal arteries. Other constituents of microorganisms or some self-antigens (e.g., oxidized lipids) might also be TLR ligands that activate the arterial dendritic cells.
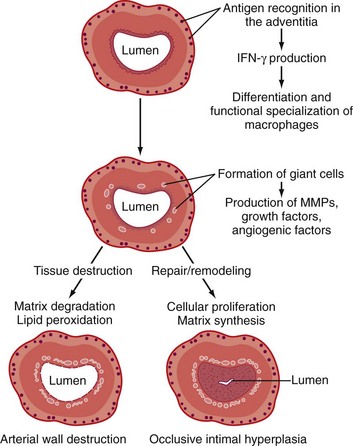
Once its TLRs have been engaged, dendritic cells differentiate from the resting to the active state and release cytokines such as IL-6 and IL-18, which recruit, activate, and retain CD4+ T cells in the blood vessel. The crucial role of dendritic cells in activating T cells and maintaining vasculitis has been demonstrated in the GCA-SCID mouse model: Depleting the dendritic cells markedly reduces the T cell infiltrate and suppresses the vasculitis. As noted, two separate populations of CD4+ T cells become activated and expand: IFN-γ producing Th1 cells and IL-17 producing Th17 cells. IFN-γ causes macrophages to migrate, differentiate, and form granulomas.34,40 Production of matrix metalloproteinases and lipid peroxidation agents by macrophages in the media results in the destruction of elastic laminae. The vessel attempts to counter the tissue destruction by elaborating a variety of growth factors including PDGF, vascular endothelial growth factor (VEGF), and TGF-β, which prompt smooth muscle cells in the media to revert from a contractile phenotype to a secretory one and migrate to the intima. Proliferation of the intimal smooth muscle cells results in occlusion of the lumen.22,32
PMR appears to result from a similar but less intense adaptive immune response in blood vessels (as evidenced by the in situ production of many inflammatory cytokines). According to this model, both PMR and GCA begin with the activation of dendritic cells at the adventitia-media border.32,41 However, the distinguishing feature of PMR is the absence of T cells producing IFN-γ. Without IFN-γ to stimulate the recruitment and differentiation of macrophages, the level of arterial inflammation in PMR remains subclinical. Thus the development of GCA appears to require both vascular dendritic cell activation and a disease-inducing repertoire of T cells.22,32,41 The constitutional symptoms of PMR and GCA are attributed to the high levels of inflammatory cytokines (e.g., IL-1, IL-6) found in the sera. Whether these serum cytokine elevations result from blood vessel inflammation alone or from some other source of inflammation is not yet clear. The attractiveness of this model is increased by its ability to explain why subsets of clinical features occur together.
Clinical Features
The mean age at onset of GCA and PMR is approximately 79 years, with a range of about 50 to 90 years of age.10,12 Younger patients with PMR have been described occasionally. Women are affected about twice as often as men.19 Although the onset of the disease is usually insidious, typically evolving over weeks or months, in one-third of cases, the disease begins so abruptly that some patients recall the very day they became ill.10,36
Giant Cell Arteritis
Classic Manifestations.
The most common manifestations of GCA are constitutional symptoms, headache, visual symptoms, jaw claudication, and PMR10 (Table 88-3). Almost all patients experience one or more constitutional symptoms including fatigue, weight loss, malaise, and fever.
Table 88-3 Symptoms of Giant Cell Arteritis
| Symptom | Frequency (%) |
|---|---|
| Headache | 76 |
| Weight loss | 43 |
| Fever | 42 |
| Fatigue | 39 |
| Any visual symptom | 37 |
| Anorexia | 35 |
| Jaw claudication | 34 |
| Polymyalgia rheumatica | 34 |
| Arthralgia | 30 |
| Unilateral visual loss | 24 |
| Bilateral visual loss | 15 |
| Vertigo | 11 |
| Diplopia | 9 |
Modified from Smetana GW, Shmerling RH: Does this patient have temporal arteritis? JAMA 287:92, 2002. Data from a review of 2475 patients reported in the literature.
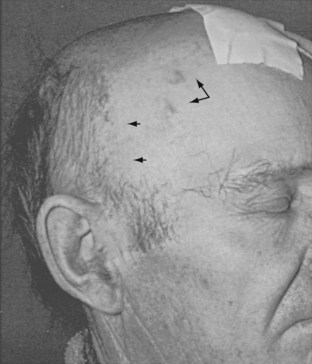
Besides constitutional symptoms, headache is the most common symptom in GCA, being present in nearly three-quarters of patients.42 The pain is typically described as boring in quality, of moderate severity, and most commonly appreciated in the temporal area. However, the description of the headache varies enormously. It can be mild to so severe that the patient seeks immediate relief by presenting to the emergency department. The pain may localize to any part of the skull including the occiput (owing to involvement of the occipital artery).10,13 The most consistent characteristic is that the patient experiences the headache as something new and unusual. In untreated patients, the headache may subside over weeks, even though the disease activity continues. Often, the headache of GCA is not associated with any particular findings on physical examination. Abnormalities of the temporal artery—including enlargement, nodular swelling, tenderness, or loss of pulse—develop in only about half of patients (Figure 88-4). Some patients note tenderness of the scalp, which can be aggravated by brushing or combing the hair.
Visual symptoms are common in GCA, especially loss of vision and diplopia. Vision loss can be unilateral or (less commonly) bilateral, transient or permanent, and partial or complete.43 Vision loss lasting more than a few hours usually does not reverse. Loss of vision often reflects an anterior ischemic optic neuropathy caused by occlusive arteritis of the posterior ciliary artery, the chief blood supply to the head of the optic nerve. The posterior ciliary artery is a branch of the ophthalmic artery (which derives, in turn, from the internal carotid artery). Less frequently, vision loss in GCA stems from a retinal artery occlusion. Regardless of the site of the culprit lesion, vision loss in GCA is usually profound, with more than 80% of patients unable to see hand waving.43 GCA patients who present with fever or other systemic symptoms are less likely to develop vision loss.44,45 One possible explanation of this protective effect of fever and other systemic manifestations is that patients with prominent systemic inflammation demonstrate more extensive angiogenesis in temporal artery biopsies.6 The angiogenesis associated with increased inflammation may result in the development of collateral circulation that reduces the chance of ischemic events.6
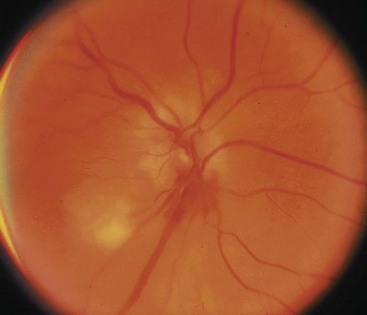
The early funduscopic appearance in the setting of blindness caused by anterior ischemic optic neuropathy is that of ischemic optic neuritis: slight pallor and edema of the optic disc, with scattered cotton-wool patches and small hemorrhages43 (Figure 88-5). Later, optic atrophy occurs. Rarely, blindness may be the initial symptom; however, it tends to follow other symptoms by several weeks or even months. Ophthalmoscopic examination in patients without eye involvement is generally normal. In most reports, the incidence of blindness is 20% or less.6,43,46 In a series of 245 patients from the modern era, 34 (14%) had some permanent loss of vision.46 In 32 of these patients, the deficit developed before glucocorticoid therapy was begun; in the other 2, vision loss occurred after therapy was started. Vision loss progressed in 3 of the 32 after therapy was initiated, and it improved in 5. At 5 years’ follow-up, among the patients who had visual deficits caused by GCA at the time glucocorticoids were started, the risk of additional loss of vision was 13% over the follow-up period. If no loss had occurred at the beginning of glucocorticoid therapy, there was only a 1% risk of new loss of vision over the subsequent 5 years.
Another potential ocular complication of GCA is ophthalmoplegia. Diplopia usually results from ocular motor nerve palsies caused by ischemia and usually resolves after therapy is started. Oculomotor nerve involvement in GCA usually spares the pupil.43 Rarely, arterial lesions cause infarction of the occipital cortex and vision loss.
Intermittent claudication may occur in the muscles of mastication (jaw claudication), the extremities, and occasionally the muscles of the tongue or those involved in swallowing.10 In the jaw muscles, the discomfort is noted, especially when chewing meat, and may involve the muscles on one side of the mandible more than those on the other. In some instances, facial artery involvement results in spasm of the jaw muscles. More marked vascular narrowing may lead to gangrene of the scalp or tongue.
Atypical Manifestations.
Approximately 40% of patients present with disease manifestations that are considered atypical33,47–49 (Table 88-4). In these patients, headache, jaw claudication, visual symptoms, and PMR do not occur or are less prominent.
Table 88-4 Atypical Manifestations of Giant Cell Arteritis
Fever occurs in up to 40% of patients with GCA, but it is usually low grade and overshadowed by other classic symptoms. However, 15% of GCA patients may present with fever of unknown origin (FUO) in which the temperature spikes are high, dominating the clinical picture.6,36 Although GCA causes only 2% of all cases of FUO, it is responsible for 16% of all such fevers in individuals older than 65 years.36 Approximately two-thirds of patients experience shaking chills and drenching sweats, features often attributed to infection or malignancy. The median temperature is 39.1° C, and the maximum 39.8° C. The white blood cell count in GCA-induced FUO is usually normal or nearly so (at least before the initiation of prednisone).
Neurologic problems occur in approximately 30% of patients.1,50 These are diverse, but most common are neuropathies and transient ischemic attacks or strokes. Hemiparesis or brain stem events are due to narrowing or occlusion of the carotid or vertebrobasilar artery. GCA preferentially involves the posterior circulation; the 3 : 2 ratio of anterior to posterior strokes and transient ischemic attacks seen in the normal population reaches nearly 1 : 1 in patients with GCA.51 Delirium, reversible dementia, and myelopathy have also been reported. However, the assignment of an exact cause to ischemic central nervous system events is often challenging, given the older population in which GCA occurs. The neuropathies of GCA include mononeuropathies and peripheral polyneuropathies and may affect the upper or lower extremities. Presumably, they are secondary to the involvement of nutrient arteries, but little pathologic documentation is available. Among the vasculitides, GCA has a nearly unique propensity for involving the C5 nerve root, resulting in loss of shoulder abduction.25 Mononeuropathies affecting the hands and feet, so typical of polyarteritis and other forms of vasculitis, develop less often in GCA.
Prominent respiratory tract symptoms occur in about 10% of patients.52 These include cough with or without sputum, sore throat, and hoarseness. When these symptoms are severe or an initial manifestation of GCA, they may direct the attention of the examining physician away from the underlying arteritis. Vasculitis may induce these symptoms by causing ischemia or hyperirritability of the affected tissues. Otolaryngeal manifestations of GCA include throat pain, dental pain, tongue pain, glossitis, and ulceration or infarction of the tongue.52,53
Clinical evidence of large artery involvement occurs in 10% to 15% of cases at presentation and in up to 27% eventually.3,38,54 Positron emission tomography (PET) studies using fluorodeoxyglucose (FDG) revealed that subclinical involvement of large arteries occurs in the vast majority of GCA patients. One PET study, for example, showed that 88% of 35 patients had increased FDG uptake in large arteries, with subclavian involvement in 7% and aortic involvement in 54%.55
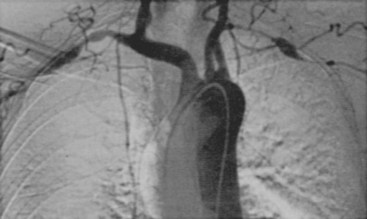
Generally, clinically evident disease can be divided into early (within a year of diagnosis) and late (years after diagnosis) stages. Usually, early disease consists chiefly of large artery stenosis resulting in upper extremity claudication; bruits over the carotid, subclavian, axillary, and brachial arteries; absent or decreased pulses in the neck or arms; and Raynaud’s phenomenon (Figure 88-6).54 Angiographic features that suggest GCA are smooth-walled arterial stenoses or occlusions alternating with areas of normal or increased caliber in the absence of irregular plaques and ulcerations, located especially in the carotid, subclavian, axillary, and brachial arteries. Late disease most frequently involves thoracic aortic aneurysm.54 The tendency for aneurysm to develop late was confirmed in one series of 41 patients in which the average time between diagnosis of GCA and recognition of this complication was 7 years.39 Thoracic aortic aneurysm is 17 times more likely to develop in patients with GCA than in persons without this disease. To place this risk in context, thoracic aortic aneurysms are twice as likely to complicate GCA as lung cancer is to result from smoking. Abdominal aortic aneurysm is also 2.4 times more common in patients with GCA.3,39 In aggregate, nearly one out of five patients (18%) with GCA develops an aortic aneurysm or dissection.54 Patients with large artery disease often do not have headache or other classic manifestations of GCA, and less than 50% have an abnormal temporal artery biopsy. Computed tomography (CT) angiography and magnetic resonance angiography (MRA) are the imaging modalities most commonly used to detect large artery disease in GCA.
In women, GCA can uncommonly present as a breast or ovarian mass.49,56 The mass lesions in these tissues result from granulomatous inflammation in and around the arteries. Angina pectoris, congestive heart failure, and myocardial infarction secondary to coronary arteritis occur rarely.
Clinical Subsets.
Studies suggest that GCA is not just one disease but rather a number of clinical subsets that are explained by the differential expression of inflammatory cytokines.57 Ischemic events including blindness, stroke, and large artery disease occur more commonly in patients who express high levels of IFN-γ and low levels of IL-6.57 In contrast, patients who produce high amounts of IL-6 are more likely to have strong inflammatory features (such as fever and constitutional symptoms) and are less likely to develop vision loss or other ischemic events.57–59
Polymyalgia Rheumatica
As in GCA, PMR patients are characteristically in good health before their disease begins.10 Systemic manifestations such as malaise, low-grade fever, and weight loss are present in more than half the patients and may be the initial symptoms. High, spiking fevers are uncommon in PMR in the absence of GCA.36 Arthralgias and myalgias may develop abruptly or evolve insidiously over weeks or months.2 Malaise, fatigue, and depression, along with aching and stiffness, may be present for months before the diagnosis is made. In most patients, the shoulder girdle is the first to become symptomatic; in the remainder, the hip or neck is involved at the onset. The discomfort may begin in one shoulder or hip but usually becomes bilateral within weeks. Symptoms center on the proximal limb, axial musculature, and tendinous attachments. Morning stiffness resembling that of RA and “gelling” after inactivity are usually prominent. If the symptoms are severe, aching is more persistent. Although movement of the joints accentuates the pain, it is often felt in the proximal extremities rather than in the joints.2 Distal joint pain and swelling occur in some cases including diffuse distal extremity swelling with pitting edema.60 Pain at night is common, and movement during sleep may awaken the patient. Muscle strength is generally unimpaired, although pain with movement makes the interpretation of strength-testing maneuvers difficult. Pain with movement also makes it difficult for patients to get out of bed or the bathtub. In the later stages of the syndrome, muscle atrophy may develop, and contracture of the shoulder capsule may result in limitation of passive and active motion.
As noted, the presence of bursal inflammation and synovitis in PMR has been described by many authors and is undoubtedly the cause of many of the findings in this condition.5 A careful examination may reveal transient synovitis of the knees, wrists, and sternoclavicular joints. The shoulders and hips are covered by heavy muscles, and minimal effusions of slight synovitis are not palpable on physical examination. Synovitis has been documented by biopsies, synovial analysis, joint scintiscans, ultrasonography, and MRI.36–38
Relationship between Polymyalgia Rheumatica and Giant Cell Arteritis
Evidence that PMR and GCA are related and should be considered different manifestations of a common disease process is abundant.10,22 The associations with age, ethnicity, geographic region, and HLA class II alleles are the same in both disorders. Moreover, both disorders involve overproduction of many of the same inflammatory cytokines. Between 30% and 50% of patients with GCA develop PMR. Approximately 10% to 15% of patients who appear to have only PMR have positive temporal artery biopsies. In the absence of symptoms of GCA (e.g., headache, jaw claudication, visual symptoms, high fever), PMR by itself does not appear to cause vision loss and responds to low doses of glucocorticoids (see later).10

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


