83 Etiology and Pathogenesis of Scleroderma
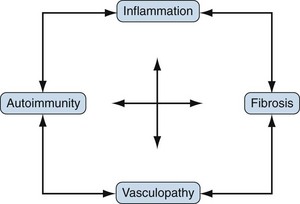
Scleroderma or systemic sclerosis (SSc) is an uncommon disease of unknown cause and complex pathogenesis. The hallmarks of SSc are (1) autoimmunity, (2) inflammation, (3) functional and structural alterations in small blood vessels, and (4) interstitial and vascular fibrosis in the skin and internal organs. This unique constellation of distinct but related pathophysiologic features, illustrated in Figure 83-1, accounts for the characteristic clinical manifestations of SSc. Early-stage disease may be dominated by inflammation and vascular injury, whereas in advanced disease fibrosis and vascular insufficiency are most prominent. However, there is enormous patient-to-patient variability in these features. Recent advances in cell and molecular biology, mouse genetic engineering, functional genomics, and genetic association studies reveal the involvement of a large number of molecules, pathways, and cell types in SSc, yielding an increasing nuanced picture of the pathogenesis. Environmental triggers in a genetically susceptible individual are thought to induce a cascade of events with early vascular injury, immune cell activation, the generation of autoimmunity, and subsequent fibroblast activation and matrix accumulation that results in chronic and progressive tissue damage. Over time, vascular insufficiency and widespread fibrosis cause disruption of vital organs, accounting for the substantial morbidity and mortality of SSc.
Etiology
Genetic Risk: Family Studies
Familial clustering of a disease is considered as evidence of inherited disease susceptibility, but such clustering might be explained by shared environmental exposures, shared genetic background, or the interaction between genes and environment.1 The risk of SSc is increased among first-degree relatives of SSc cases compared with the general population. In one U.S. study, the relative risk of SSc among first-degree relatives of cases was 13, with a rate of 1.6% compared with 0.026% in the general population, identifying a family history of SSc as the strongest known risk factor.2 The only twin study of SSc to date reported a relatively small disease concordance rate (4.7%), although the concordance rate for positive antinuclear antibody (ANA) was 90% for monozygotic (identical) twins and 40% for dizygotic (fraternal) twins.3 Raynaud disease and pulmonary fibrosis show increased prevalence in pedigrees of SSc patients.4 Moreover, autoimmune diseases among first-degree family members of SSc patients have been reported in up to 36% of cases, with hypothyroidism, hyperthyroidism, rheumatoid arthritis, and systemic lupus erythematosus (SLE) most common.
Genetic Association Studies: Immune Susceptibility Genes for Scleroderma
The past decade has witnessed rapid progress in delineating genetic susceptibility factors in SSc. Genetic association studies using candidate gene approaches and, more recently, genome-wide association (GWA) studies have been performed in large multinational patient cohorts. The major histocompatibility complex (MHC) is the dominant genetic region implicated in autoimmune disease, although the role of specific human leukocyte antigen (HLA) alleles in pathogenesis remains unknown. The interpretation of HLA associations is complicated by the extensive linkage disequilibrium of risk haplotypes. Specific HLA alleles have long been known to be associated with SSc and specific autoantibodies. For instance, a case-contol study of SSc revealed strong associations with HLA DRB1*1104, DQA1*0501, and DQB1*0301 haplotypes.5 Candidate gene approaches typically look for changes in single nucleotides (single nucleotide polymorphisms [SNPs]), the most common form of deoxyribonucleic acid (DNA) variation. Non-HLA susceptibility genes associated with SSc include the protein tyrosine phosphatase nonreceptor 22 (PTPN22), which has been associated with SLE, myasthenia gravis, vitiligo, and Addison’s disease; interleukin (IL)-1β and NLRP1, an inflammasome scaffold that promotes pro-IL-1β maturation and processing; and interferon regulatory factor 5 (IRF5), a transcription factor in the Toll-like receptor (TLR) pathway that mediates type I interferon (IFN) induction and is associated with SLE, as well as SSc and interstitial lung disease. The association of IRF5 with SSc is particularly interesting, in light of the role of IFN type I in immune responses. Table 83-1 summarizes SSc susceptibility genes identified to date. In addition to classic SNPs, informative genetic polymorphisms in SSc include variations of copy numbers, rare allelic variants, and epigenetic changes. The identification of these genetic associations will require in-depth analysis using deep-sequencing technologies.
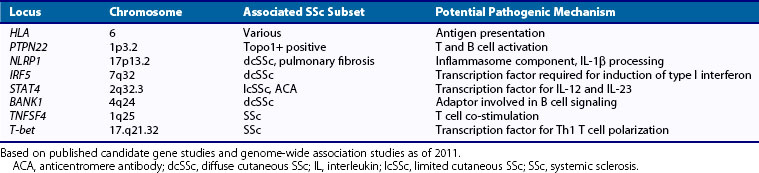
Other Candidate Genes and Genome-wide Association Studies
Several GWA studies in large and ethnically diverse populations are currently under way. These unbiased approaches have the advantage over candidate gene studies in that they are driven by discovery rather than a priori hypotheses. The first large-scale SSc GWA study analyzed 300,000 SNPs in a cohort of 2296 white cases and 5014 healthy controls.6 Statistically significant associations were found with SNPs in the HLA region, as well as IRF5, TNFAIP3, and CD247, a gene implicated in T cell signaling and also associated with susceptibility to SLE. Although genetic association studies represent a rapidly evolving area of research, the results to date can be summed up as follows: (1) Genetic variants associated with SSc susceptibility are involved in innate and adaptive immune responses, and (2) they are shared with SLE and other autoimmune diseases. It is worth noting that important associations can be missed by GWA studies using current technologies. Moreover, despite the wealth of emerging information, the genetic associations discovered to date are of relatively modest magnitude, with odds ratios that are generally less than 1.5. This finding points to the potential importance of gene-gene interactions (epistasis), particularly for genes within the same molecular pathways, and gene-environment interactions. A current challenge in SSc research is how to handle the large volume of new information emerging from GWA studies. Moreover, it will be important to delineate how genes shared among diverse autoimmune conditions contribute to disease-specific phenotypes. Shedding light on this important problem will require large collaborative studies involving phenotypically well-characterized populations of varied ethnic background and meta-analyses.
Infectious Agents: Viruses
Along with exposure to certain environmental and occupational agents and drugs, viruses such as human cytomegalovirus (hCMV) and parvovirus B19 have been implicated as potential triggers for SSc. Patients with SSc have anti-hCMV antibodies directed against the UL83 and UL94 protein epitopes on hCMV. Anti-UL94 antibodies can induce endothelial cell apoptosis and fibroblast activation, suggesting a direct role for antiviral antibodies in tissue damage. Antitopoisomerase-I can cross-react with hCMV-derived proteins, implicating molecular mimicry as a mechanism linking hCMV infection and SSc.7 Cytomegalovirus is implicated in the pathogenesis of allograft vasculopathy, a complication of organ transplantation characterized by vascular neointima formation and smooth muscle cell proliferation strikingly reminiscent of the obliterative proliferative vasculopathy seen in SSc. In dermal fibroblasts, hCMV stimulates synthesis of the profibrotic growth factor connective tissue growth factor (CTGF, CCN2) in vitro.8 Evidence of human parvovirus B19 infection has also been reported in patients with SSc.
Environmental Exposures, Dietary Supplements, Drugs and Radiation
Although reports of putative geographic clustering suggest a role for shared environmental exposures, careful investigations have generally failed to substantiate apparent clusters of SSc. On the other hand, well-documented epidemic outbreaks of SSc-like illnesses with acute onset and chronic course have been reported. One such illness, called the toxic oil syndrome, occurred in Spain and was linked to the ingestion of contaminated rapeseed cooking oils.9 In the United States, dietary supplements containing L-tryptophan were implicated in an explosive outbreak of the eosinophilia-myalgia syndrome (EMS) in 1989.10 The EMS epidemic subsided following the ban on L-tryptophan, but sporadic cases of EMS following ingestion of L-tryptophan and other food supplements continue to be reported. Although scleroderma-like skin fibrosis was a prominent manifestation of these apparently de novo toxicoepidemic syndromes, along with multisystem involvement, chronic course, and autoimmunity, the associated clinical, histopathologic, and laboratory features clearly differentiate them from SSc.11 The frequency of SSc appears to be increased among men with occupational exposure to silica dust. This was recently confirmed by a meta-analysis of 16 observational studies, with risk estimates as high as 15.12 Other exposures linked with SSc include polyvinyl chloride, toluene, xylene, trichloroethylene, and organic solvents. Additional reports alleged an association between SSc and environmental exposures to pesticides, hair dyes, and industrial fumes. Although exposure to cigarette smoke is known to increase the risk of multiple autoimmune diseases, there is no evidence to date implicating it as a risk factor for SSc.
Certain drugs have been implicated in SSc-like illnesses. The best studied is the anticancer drug bleomycin, which induces skin and lung fibrosis in the mouse (see later). Other potentially implicated drugs include pentazocine, the taxenes docetaxel and paclitaxel, and cocaine. The use of appetite suppressants has been linked to the development of pulmonary arterial hypertension (PAH). The occurrence of SSc in women following cosmetic breast augmentation with silicone implants raised significant concern regarding a possible causal association.13 Subsequent large-scale epidemiologic surveys and a meta-analysis, however, failed to confirm an increased risk of SSc or of other connective tissue diseases among women with silicone breast implants.14 Radiation treatment for malignant neoplasms has been linked with the onset of de novo SSc, as well as exacerbation of tissue fibrosis in patients with pre-existing SSc.15 Some of the environmental agents and drugs that have been linked with the development of SSc are listed in Table 83-2.
Table 83-2 Environmental Agents and Drugs Implicated in Scleroderma-like Syndromes
| Chemicals |
| Drugs |
| Dietary Supplement/Appetite Suppressants |
Microchimerism
Healthy women harbor immunologic stem cells of fetal origin that persist for many years following pregnancy, a condition called microchimerism. Some studies found that the number of circulating fetal cells is elevated in women with SSc compared with healthy women.16,17 Moreover, cells with a male chromosome, presumably from a prior pregnancy with a male fetus, have been detected in affected organs from women with SSc. It has been speculated that persistence of fetal cells in SSc may be linked to the development of the disease through a graft-versus-host response triggered by the fetal cells or through a maternal (auto)immune response against the fetal cells.
Pathology
General Features
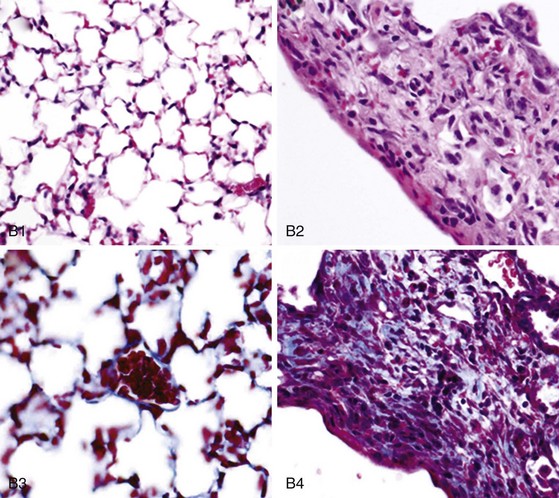
The pathologic hallmarks of SSc are a noninflammatory proliferative/obliterative vasculopathy affecting small arteries and arterioles in multiple vascular beds and interstitial and vascular fibrosis, most prominent in the skin, lungs, and heart.18 Inflammation is generally absent in long-standing SSc, but in early-stage disease inflammatory cell infiltrates in many organs may be prominent. In the skin, the infiltrates are located predominantly around blood vessels and in the reticular dermis and are composed primarily of CD4+ T lymphocytes and monocytes, whereas in the lungs, cellular infiltrates consist predominantly of CD8+ T lymphocytes.
Vascular Pathology
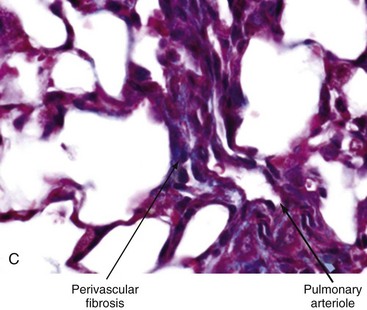
Vascular injury and activation are the earliest and possibly primary events in SSc. Histopathologic evidence of vascular damage is present before fibrosis and can be detected in involved and uninvolved skin, indicating a generalized process.19 Raynaud phenomenon and other vascular manifestations typically precede other manifestations of SSc. Additional signs of vasculopathy include cutaneous telangiectasia; nailfold capillary changes (giant capillaries, hemorrhages, and avascular areas); PAH; digital tip pitting; gastric antral vascular ectasia (also called watermelon stomach); and scleroderma renal crisis. The most characteristic histopathologic finding in the small and medium-sized arteries is bland intimal proliferation (Figure 83-2). Intimal hypertrophy, a finding that SSc shares with chronic allograft arteriopathy, is thought to result from proliferation and migration of myointimal cells and local accumulation of collagen.20 The vascular basement membranes are thickened and reduplicated. These changes are most prominent in blood vessels of the heart, lungs, kidneys, and gastrointestinal tract. A systematic survey of SSc skin biopsies revealed a marked reduction in the number of capillaries (rarefaction) and loss of vascular endothelial cadherin, a molecule required for vascular tube formation.21 Remarkably, this study showed that clinical improvement following high-dose immunosuppressive therapy was accompanied by capillary regeneration in the skin.
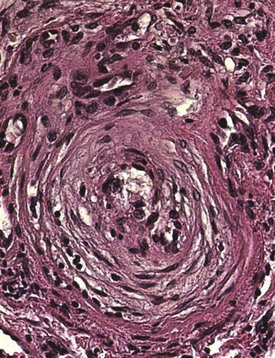
Impaired fibrinolysis, increased levels of von Willebrand factor, and ongoing platelet aggregation are prominent. Endothelial cell injury itself causes further platelet aggregation, release of platelet-derived growth factor (PDGF) and endothelin-1 (ET-1), and endothelial cell apoptosis.22 Vasculitic lesions and immune complex deposition in the vessel walls are uncommon. In late stages of the disease, extensive fibrin deposition and perivascular fibrosis cause progressive luminal occlusion, and eventually there is striking paucity of small blood vessels and capillaries in lesional tissue.23 Loss of vascular supply leads to chronic tissue hypoxia. Widespread proliferative/obliterative vasculopathy of small and medium-sized arteries and capillary rarefaction are the pathologic hallmarks of all forms of SSc. In patients with SSc-associated PAH, pulmonary arteriolar intimal proliferation and evidence of veno-occlusive disease are prominent, whereas in contrast to idiopathic PAH, plexogenic arteriopathy does not occur.24
Organ-Specific Pathologic Findings
Skin
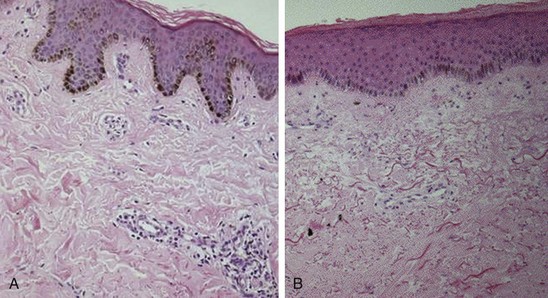
Fibrosis of the skin, the hallmark of SSc, is accompanied by marked expansion of the dermis with obliteration of the hair follicles, sweat glands, and sebaceous glands and other skin appendages. Collagen fiber accumulation is most prominent in the reticular (deep) dermis and progressively invades the subjacent adipose layer with entrapment of fat cells. Early-stage SSc skin biopsies reveal dermal edema and perivascular infiltrates composed of T lymphocytes and monocytes (Figure 83-3). Less commonly, mast cells and eosinophils may also be detected.25,26 The proportion of α–smooth muscle actin–positive myofibroblasts, a mesenchymal cell type that is intermediate between fibroblasts and contractile smooth muscle cells and plays a major role in fibrogenesis, is increased in the lesional skin.27 With disease progression, the skin undergoes atrophy with loss of epidermal-dermal ridges and effacement of the rete pegs reminiscent of the changes seen in aging skin. The fibrotic dermis is largely acellular and contains dense accumulation of compact hyalinized collagen bundles, fibronectin, hyaluronic acid, and other structural proteins. Sweat glands and eccrine glands atrophy with loss of periglandular adipose tissue. The subcutaneous adipose layer is obliterated. In a double-blind study of 45 SSc skin biopsies, the histologic grade of skin fibrosis was found to closely correlate with the extent of clinical skin involvement.28 Reduction in the number of dermal lymphatic vessels, which can be marked, contributes to interstitial fluid accumulation and edema.29 Paucity of dermal capillaries (rarefaction) is associated with chronic tissue hypoxia that induces angiogenic factors such as vascular endothelial growth factor (VEGF). Evidence of tissue hypoxia can even be found in clinically uninvolved, apparently “normal” skin.30
Biochemical analysis shows that the collagens in the fibrotic dermis are normal, and relative proportions of the main fibrillar collagens (type I and type III) are comparable with those of normal skin.31 In contrast, the minor nonfibrillar type VII collagen, normally restricted to the dermal-epidermal basement membrane zone, is abundant throughout the lesional dermis. The enzymes mediating post-translational collagen modification such as lysyl hydroxylase (PLOD2) are elevated, resulting in an increase in aldehyde-derived collagen cross-links, which may account for the dense sclerotic nature of the fibrotic dermis.32
Genome-wide expression profiling of lesional skin using DNA microarray technology provides an increasingly clearer understanding of the activation events that underlie fibrosis. Results from several studies reveal strikingly altered gene expression patterns in SSc skin biopsies compared with healthy controls. Remarkably, clinically involved and uninvolved skin appear to be indistinguishable in terms of their gene expression profiles. Many genes involved in ECM homeostasis and in transforming growth factor-β (TGF-β), CCN2, IL-13, and Wnt signaling pathways show elevated expression.33,34 Furthermore, a number of genes reflecting a bone/cartilage phenotype are elevated in the skin. A particularly intriguing finding from these studies is that skin biopsies from different individuals show marked heterogeneity at the level of their “molecular signature,” with at least five distinct and reproducible patterns identified to date.35
Lungs
In early-stage SSc, the lungs show patchy infiltration of the alveolar walls with lymphocytes, plasma cells, macrophages, and eosinophils (Figure 83-4). At this stage, bronchoalveolar lavage fluid contains elevated proportions of inflammatory leukocytes. With progression, interstitial lung fibrosis and vascular damage predominate, often coexisting within the same lesions. Intimal thickening of the pulmonary arteries, best seen with elastin stain, underlies PAH and at autopsy is often associated with multiple pulmonary emboli and myocardial fibrosis.
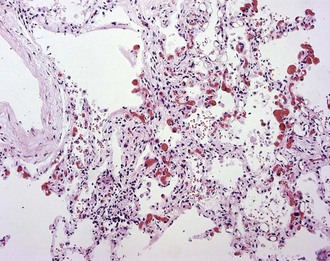
Lung fibrosis in SSc is characterized by expansion of the alveolar interstitium due to accumulation of collagens and other connective tissue proteins. The typical histologic pattern is nonspecific interstitial pneumonitis (NSIP), a form of interstitial lung disease characterized by mild to moderate interstitial inflammation, type II pneumocyte hyperplasia, and fairly uniform distribution of fibrosis. Less commonly, SSc is associated with the usual interstitial pneumonia (UIP) pattern that is characterized by scattered fibroblastic foci and patchy distribution of fibrosis and has a worse prognosis.36 Progressive alveolar septal thickening ultimately results in obliteration of the air spaces and honeycombing, as well as consequent loss of pulmonary blood vessels. This process impairs gas exchange and contributes to worsening of pulmonary hypertension. Extensive pulmonary fibrosis may also predispose to primary lung carcinoma.
Gastrointestinal Tract
Prominent pathologic changes can occur at any level from the mouth to the rectum. The esophagus is virtually always affected, with fibrosis in the lamina propria, submucosa, and muscular layers and characteristic vascular lesions.37 Replacement of the normal intestinal architecture results in disordered peristaltic activity, gastroesophageal reflux and small bowel dysmotility, pseudo-obstruction, and bacterial overgrowth. Chronic gastroesophageal reflux is complicated by esophageal inflammation, ulcerations, and stricture formation. Up to one-third of SSc patients with severe gastroesophageal reflux develop Barrett’s esophagus, characterized by metaplasia of the normal squamous lining of the esophagus into columnar epithelium.38 Because Barrett’s metaplasia is a premalignant lesion associated with a greater than 30-fold increased risk of adenocarcinoma, patients with Barrett’s esophagus need ongoing monitoring for dysplasia and adenocarcinoma.
Kidneys
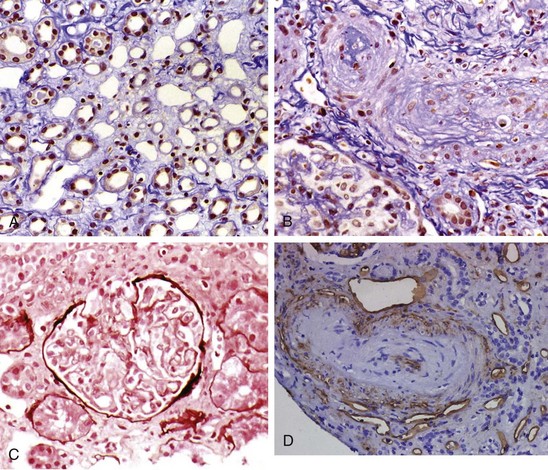
In the kidneys vascular lesions predominate, and glomerulonephritis is rare except in overlap syndromes. Chronic renal ischemia is associated with shrunken glomeruli and other ischemic changes. Patients with acute scleroderma renal crisis show a thrombotic microangiopathy that is indistinguishable from other forms of malignant hypertension.39 Histopathologic changes are most prominent in the small interlobular and arcuate renal arteries, which show reduplication of elastic lamina, marked intimal proliferation (onion skinning), and fibrinoid necrosis of the arteriolar walls.40 Similar pathologic changes have also been reported in SSc patients who do not have renal crisis. Intimal thickening leads to severe narrowing and total obliteration of the lumen, often with microangiopathic hemolysis. Tubular changes such as flattening and degeneration of tubular cells occur secondary to vascular insufficiency. The clinical picture of scleroderma renal crisis may resemble thrombotic thrombocytopenic purpura (TTP). However, reduced to absent levels or activity of von Willebrand factor cleaving protease (ADAMTS13) has not been reported in scleroderma renal crisis. Figure 83-5 shows the characteristic histologic features associated with scleroderma renal crisis.
Heart
At autopsy, evidence of cardiac involvement is found in up to 80% of patients with SSc.41 Modest pericardial effusions are common; occasionally fibrosis and constrictive pericarditis may occur. A characteristic pathologic finding is myocardial contraction band necrosis, which is thought to reflect repeated episodes of ischemia-reperfusion due to “myocardial Raynaud phenomenon.”42 Significant interstitial and perivascular fibrosis in the heart may occur in the absence of clinically evident heart involvement. Skeletal muscle myositis in SSc is occasionally accompanied by acute myocarditis.43
Pathologic Findings in Other Organs
Fibrosis of the thyroid glands is common. Broad bands of fibrous tissue are seen in the thyroid gland, with atrophy and obliteration of the follicles, in the absence of inflammation. Abnormal thyroid function tests and antithyroid antibodies are common. Erectile dysfunction is frequent and may be a presenting manifestation of the disease in men. Pathologic examination shows extensive proliferative/obliterative changes in the penile blood vessels.44 Fibrosis of the salivary and lacrimal glands in the absence of inflammation can occur and may be associated with Sjögren’s syndrome. Synovial biopsies show fibrosis and characteristic vascular changes in the small arterioles.45
Animal Models of Scleroderma
Animal models of human disease are indispensible research tools to facilitate the understanding of complex disease states. Such models are used to identify the cellular and molecular components of pathologic process, discover potential therapeutic targets, and develop and evaluate novel treatment strategies. A large number of putative SSc models have been reported, but to date none fully reproduce each of the four cardinal features of the disease: obliterative and proliferative microangiopathy, autoimmunity, inflammation, and fibrosis.46 However, as illustrated in Figure 83-6, particular animal models recapitulate selected disease features. Broadly speaking, current mouse models can be divided into four types: (1) naturally occurring disease models, in which spontaneous mutations are associated with a genetically transmitted scleroderma-like phenotype such as tight skin (Tsk1/+ mouse); (2) induced models in which the scleroderma phenotype is elicited by chemical exposures or by manipulation of the immune system (bleomycin-induced skin and lung fibrosis); (3) transplantation of HLA-mismatched bone marrow cells resulting in chronic sclerodermatous graft-versus-host disease; and (4) genetic manipulations giving rise to engineered mouse strains with heritable scleroderma-like traits.
Heritable Animal Models of Scleroderma
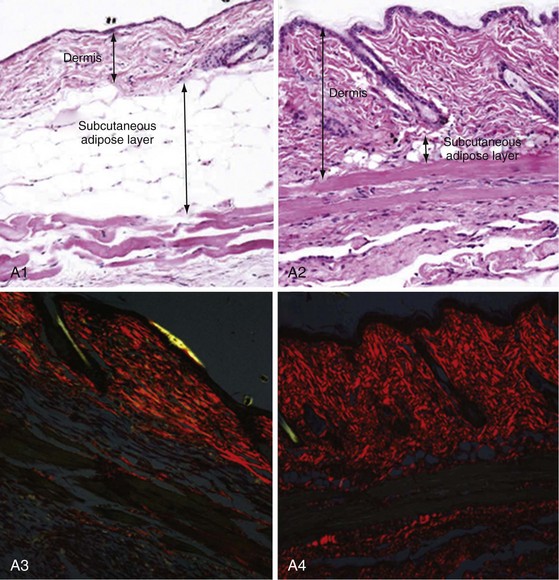
The Tsk1/+ mouse is characterized by diffuse skin thickening and tethering to the underlying subcutaneous tissue. Mice homozygous for the Tsk1 mutation die in utero at 8 to 10 days of gestation. However, heterozygous mice (Tsk1/+) survive and develop tight skin. In contrast to SSc, Tsk1/+ mice show prominent subcutaneous hyperplasia but relatively unremarkable dermis.47 Furthermore, the lungs of Tsk1/+ mice show emphysematous changes rather than fibrosis, and microangiopathy has not been reported. Although inflammation is uncommon, Tsk1/+ mice develop autoantibodies directed against topoisomerase-I. The Tsk1 mutation is now known to be an intragenic tandem duplication fibrillin-1 gene.48 Fibrillin-1 is a large ECM protein that is widely distributed in microfibrils, and in addition to its structural role also modulates the latency and activation of TGF-β.49 Fibrillin-1 gene mutations are associated with Marfan’s disease with multiple tissues showing activation of TGF-β. The fibrillin-1 duplication mutation in Tsk1/+ mice gives rise to an abnormally large 450-kD protein. No corresponding fibrillin-1 mutations have been demonstrated in patients with SSc. Although it has been hypothesized that accumulation of the abnormally large mutant fibrillin-1 destabilizes the matrix50 or perturbs the homeostatic control of TGF-β latency, the precise mechanisms linking the Tsk1/+ mutation in fibrillin-1 to the development of cutaneous hyperplasia are unknown.
Another animal model of scleroderma is the Tsk2 mouse that spontaneously develops scleroderma-like skin changes by 3 to 4 weeks of age.51 In contrast to Tsk1/+ mice, Tsk2/+ mice have fibrotic dermis with extensive infiltration with mononuclear inflammatory cells and show evidence of autoimmunity. The Tsk2 mutation, originally induced in normal mice by exposure to ethylnitrosourea, is located on chromosome 1 and is inherited as an autosomal dominant trait, although the underlying molecular defect has not yet been identified.
Inducible Animal Models of Scleroderma
Chronic skin and lung fibrosis (Figure 83-7) can be induced in BALB/c or C57 mice by subcutaneous injections of bleomycin or oxidative agents. Bleomycin is an antitumor chemotherapy drug that has long been recognized to be complicated by pulmonary fibrosis. The sequence of bleomycin-induced histopathologic changes in mice closely resembles those seen in SSc: early and self-limited mononuclear cell infiltration and increased expression of cytokines such as TGF-β, IL-4, IL-6, IL-13, and monocyte chemotactic protein-1 (MCP-1), followed by the appearance of dermal fibrosis with excessive collagen deposition and accumulation of α–smooth muscle actin–positive myofibroblasts.52 Bleomycin-induced fibrosis may be linked to reactive oxygen species (ROS) generation, as well as direct activation of innate immunity via TLR2. In contrast to SSc, bleomycin-induced mouse scleroderma is not associated with either microangiopathic changes or autoantibodies and skin fibrosis is limited in extent and duration. Nevertheless, in light of its reproducibility, relative strain independence, and ease of induction, this mouse model is widely used for investigating specific molecules and pathways in the development and treatment of fibrosis. Injection of oxidative agents (such as hydroxyl radicals or hypochlorite) into the skin in BALB/c mice induces skin and lung fibrosis, as well as the appearance of SSc-specific autoantibodies.53 The pathologic changes are linked to the generation of hydrogen peroxide and other ROS. Subcutaneous injection of TGF-β induces granulation tissue and only transient fibrosis, but simultaneous injection of CTGF along with TGF-β results in persistent fibrosis, suggesting that CTGF is required for sustaining the fibrotic response. However, adenovirus-mediated delivery of constitutively active TGF-β receptor I is sufficient to induce local dermal fibrosis. Transplantation of bone marrow or spleen cells into sublethally irradiated minor histocompatibility locus–mismatched recipient mice results in sclerodermatous graft-versus-host disease. This mouse model displays interstitial and perivascular firbosis in the skin and lung and autoimmunity.54 Skin fibrosis is preceded by mononuclear cell infiltration with elevated TGF-β and chemokine expression.
Genetic Manipulations in Mice Giving Rise to Scleroderma-like Phenotypes
Mouse strains with genetic gain-of-function or loss-of-function modifications resulting in spontaneous scleroderma-like phenotypes have been created. These transgenic, knockin, and knockout mice provide robust novel experimental tools in scleroderma research. Mouse strains with constitutive or inducible expression of TGF-β signaling in fibroblasts recapitulate key clinical, histologic, and biochemical features of SSc and provide support for the role of perturbed TGF-β signaling in pathogenesis.55,56 Other promising transgenic models of SSc include mice overexpressing CTGF, PDGF receptor (PDGFR)-α, Wnt10b, and Fra-2 and mice null for caveolin-1, relaxin, Fli-1, and fetuin A.57 Scleroderma-like fibrotic, vascular, and calcific changes in the skin, as well as in some cases the lungs, develop spontaneously in these mice. Other genetically engineered mice show increased sensitivity to the induction of fibrosis. Examples include T-bet null and VEGF null mice. These emerging mouse models provide new exciting research opportunities for the discovery of molecules and pathways underlying the pathogenetic features of SSc.
Pathogenesis
Integrated Overview
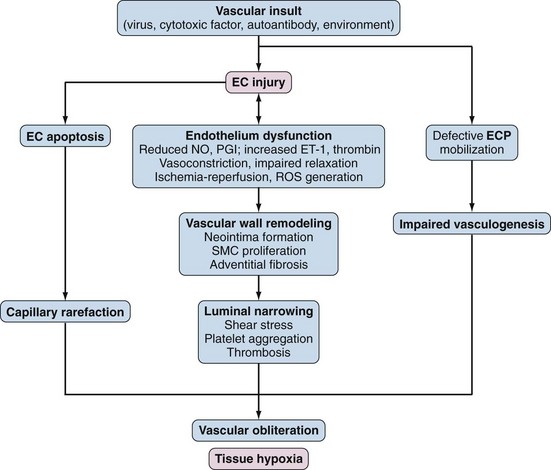
The pathogenesis of SSc is complex, and existing animal models capture only some of its diverse pathologic and clinical attributes. An integrated view of pathogenesis must integrate the cardinal features of SSc: vascular injury and damage, inflammation with activation of the innate and adaptive arms of the immune system, and fibroblast activation resulting in generalized interstitial and vascular fibrosis. Although evidence for each can be found in each SSc patient, the relative individual contribution of these processes to the clinical manifestations varies from one patient to another. As illustrated in Figure 83-1, complex and dynamic interplay between these distinct processes is thought to be responsible for initiating, amplifying, and sustaining tissue damage in SSc.58
Vasculopathy
Vascular injury is likely to be the initiating and proximal event in SSc (see Figure 83-5). Evidence of vascular involvement is early and widespread, and its progression over time is associated with significant clinical sequelae. The presence of nailfold microvascular changes, detected by capillaroscopy, in an individual with isolated Raynaud phenomenon identifies an elevated risk of progression to SSc, indicating that microvasculopathy precedes other clinical manifestations of the disease.
Vascular Injury and the Activated Endothelium
The initial vascular insult is triggered by circulating factors such as (unidentified) cytotoxic molecules or T cell–derived proteolytic granzymes.59 Other potential causes of vascular injury include antiendothelial cell autoantibodies, vasculotropic viruses, inflammatory cytokines, ROS, and other forms of environmental stress. Vascular injury causes endothelial cell activation, with increased expression of vascular endothelial cell adhesion molecule-1 and E-selectin, altered secretion of vasoactive mediators, platelet activation, and activation of the thrombotic and fibrinolytic cascades.60 In the injured arterioles and capillaries, activated endothelial cells may undergo transdifferentiation to mesenchymal cells via a process termed endothelial-mesenchymal transition. This process driven by TGF-β and Notch is associated with loss of endothelial markers such as CD31 and progressive acquisition of mesenchymal markers such as α–smooth muscle actin. Although endothelial-mesenchymal transition has been documented in cancer, recent studies identify endothelial cell-derived fibroblasts in cardiac and pulmonary fibrosis, suggesting that this form of endothelial cell plasticity may also play a role in SSc. Platelet activation is a prominent early feature of vascular injury in SSc and is associated with the release of thromboxane A2, PDGF, and TGF-β, which potentiate vasoconstriction and also contribute to fibroblast activation and myofibroblast transdifferentiation. Pericytes, which are smooth muscle–like structural cells found in the walls of small blood vessels, show marked hyperplasia in lesional skin from patients with early-stage SSc and express the surface marker Thy-1 (CD90) and receptors for PDGF.61,62
Functional abnormalities of the vascular endothelium include impaired production of and responsiveness to endothelium-derived vasodilatory factors such as nitric oxide (NO), thrombomodulin, calcitonin gene-related peptide, and prostacyclins. The ensuing imbalance of vasodilators and vasoconstrictors impairs blood flow responses. Recurrent episodes of ischemia-reperfusion create oxidative stress with the generation of H2O2 and other ROS. Damaged microvessels show increased vascular permeability and enhanced transendothelial leukocyte migration. Platelets are exposed to subendothelial structures, which further aggravates platelet aggregation. Activated endothelial cells release the extremely potent vasoconstrictor ET-1, which promotes leukocyte adhesion, vascular smooth muscle cell proliferation, and fibroblast activation (Figure 83-8). Levels of ET-1 are elevated in the blood and in bronchoalveolar lavage fluids from patients with SSc.63 Ultimately fibrinolytic and coagulation cascades are activated, culminating in intravascular fibrin deposition and thrombosis.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


