95 Clinical Features and Treatment of Gout
• Elevated serum urate concentration (hyperuricemia)
• Recurrent attacks of acute arthritis in which monosodium urate monohydrate crystals are demonstrable in synovial fluid leukocytes
• Aggregates of sodium urate monohydrate crystals (tophi) deposited chiefly in and around joints, which sometimes lead to deformity and crippling
• Renal disease involving glomerular, tubular, and interstitial tissues and blood vessels
These manifestations can occur in various combinations.1,2
Epidemiology
Key Points
Serum urate levels are low in childhood and increase in men at puberty and in women at menopause.
Hyperuricemia is fairly common, with prevalence ranging between 2.6% and 47.2% in various populations.3,4 A variety of factors appears to be associated with high serum urate concentrations. In adults, serum urate levels correlate strongly with the serum creatinine and urea nitrogen levels, body weight, height, age, blood pressure, and alcohol intake.5 In epidemiologic studies, body bulk (as estimated by body weight, surface area, or body mass index) has proved to be one of the most important predictors of hyperuricemia in people of many different races and cultures, with rare exceptions.6–8
Serum urate concentrations vary with age and sex. Children normally have a concentration in the range of 3 to 4 mg/dL because of high renal uric acid clearance.9 At puberty, serum urate concentrations increase by 1 to 2 mg/dL in males, and this higher level is generally sustained throughout life. In contrast, females exhibit little change in the serum urate concentration until menopause, when concentrations increase and approach those seen in adult men. The mechanism of lower serum urate levels in women is a consequence of sex hormones and is related to a higher fractional excretion of urate secondary to lower tubular urate postsecretory reabsorption.10
The incidence of gout varies among populations, with an overall prevalence ranging from less than 1% to 15.3%.5 This upper limit appears to be increasing.11,12 The prevalence increases substantially with age and with increasing serum urate concentration. The annual incidence rate of gout is 4.9% for urate levels greater than 9 mg/dL, 0.5% for values between 7 and 8.9 mg/dL, and 0.1% for values less than 7 mg/dL.13 For serum urate values greater than 9 mg/dL, the cumulative incidence of gout reaches 22% after 5 years.
Environmental Factors
Key Points
Certain foods clearly promote hyperuricemia and gout including alcohol, seafood, and red meat.
The consumption of some foods may be protective, especially milk and yogurt.
An association between alcohol consumption and gout has been recognized for centuries. The risk of developing gout varies by the type of alcohol ingested.14 Beer, which is purine rich, carries the highest risk; this risk is substantially greater than that for liquor. Moderate wine drinking does not increase the risk of gout. The quantity of alcohol also strongly correlates with gout. Compared with men who did not consume alcohol, the relative risk of gout was 1.32 for an alcohol intake of 10 to 14.9 g/day, 1.49 for 15 to 29.9 g/day, 1.96 for 30 to 49.9 g/day, and 2.53 for 50 g/day and higher, with calculations based on 12.8 g of alcohol per 12 oz serving of regular beer, 11.3 g per 12 oz. serving of light beer, 11 g per 4 oz. serving of wine, and 14 g per shot of liquor.
Diet also influences hyperuricemia and gout. Serum urate levels increase with meat or seafood intake and decrease with dairy intake.15 Men in the highest quintile of seafood consumption have a 51% higher risk of developing gout, and those in the highest quintile of meat intake have a 41% higher risk. However, consumption of oatmeal and purine-rich vegetables (e.g., peas, mushrooms, lentils, spinach, cauliflower) is not associated with an increased risk for gout. The consumption of milk one or more times a day or yogurt consumption at least once every other day is associated with lower serum urate levels.
Genetics
Since antiquity, gout has been recognized as a familial disorder. The familial incidences reported range from 11% to 80%.16 In two large series, one English and one American, about 40% of gouty subjects gave a positive family history of gout. These wide discrepancies may be attributed in part to variations in diligence and pursuit of genealogic data. When all available data are considered, they suggest that serum urate concentrations are controlled by polygenic traits. Several rare forms of hyperuricemia and gout such as hypoxanthine phosphoribosyltransferase deficiency, phosphoribosyl-1-pyrophosphate synthetase overactivity, and familial hyperuricemia nephropathy have a clear genetic basis, most presenting in childhood or early adulthood17 (see later).
But the vast majority of gout patients do not have one of these discrete inborn errors of metabolism as explanations for their disease. In most gout patients, the mechanism of hyperuricemia is simply inefficient renal excretion of uric acid.18,19 It stands to reason then that some of the familial predisposition to gout is related to inherited variations in renal urate handling. With the recent advances in our understanding of the transport of uric acid in the renal proximal tubule, some of this variability is being clarified. Genome-wide association studies (GWASs) have identified genetic variations of the SLC2A9/GLUT9 and ABCG2 genes as important determinants of serum urate levels (see Chapter 94).20 Curiously, the data for the role of polymorphisms in URAT1 (SLC22A12), a key urate transporter involved in renal proximal tubule urate reabsorption, are thus far inconsistent.21–25 Glucose transporter 9 (GLUT9, SLC2A9) is an electrogenic hexose transporter whose splicing variants mediate reabsorption of uric acid, along with glucose and fructose, at the renal proximal tubule epithelial cell, first at the apical membrane, then through the basolateral membrane, and on into the circulation.26–29 Polymorphisms in SLC2A9 are associated with a lower serum uric acid, with stronger effects seen in women, possibly accounting for 0.5% to 2% of the variance in serum urate concentration in men and 3.4% to 8.8% in women.22,27,30–32 In five of six populations studied by GWAS, SLC2A9 polymorphisms were associated with a decreased incidence of self-reported gout, particularly in women.27,30,31 Indeed, three patients with renal hypouricemia have been identified with loss-of-function mutations in SLC2A9.28,33 The implication is that certain polymorphisms in SLC2A9 enhance renal uric acid excretion, lower serum urate levels, and are therefore protective against gout, particularly in women.
A gene product of ABCG2, human adenosine triphosphate (ATP)-binding cassette, subfamily G, 2, appears to be a secretory urate transporter located in the renal proximal tubule apical border.34 Polymorphisms in ABCG2 are associated with a higher serum urate concentration in humans, with a more potent influence in men, potentially accounting for 1.6% to 2.1% of the variance in serum urate concentration in men and 0.5% to 0.8% in women.23,31 One ABCG2 polymorphism, rs2231142, has been associated with a higher incidence of gout only in males (odds ratio [OR], 2.03 for self-reported gout).31 In a transfection model in Xenopus oocytes, the common mutation in human ABCG2 encoded by rs2231142, Q141K, resulted in a 53% decrease in urate transport compared with the wild-type gene product.34 The implication is that certain polymorphisms in ABCG2 result in reduced proximal tubule urate secretion, increased serum urate concentrations, and a higher incidence of gout, at least in men.
Because the impact of comorbidities and environmental influences such as diet and lifestyle is so significant in gout, it remains to be seen whether risk contribution from genetic variation is clinically relevant, either directly or due to an unexpected interaction with these other factors. An example of the latter is the potential influence the urate-fructose/glucose exchange transporter SCL2A9 polymorphisms may have on the well-described association of heavy consumption of soft drinks with elevated serum urate levels and gout.35–38
Clinical Features
Key Points
The first gout attack generally occurs at age 40 to 60 in men and after age 60 in women.
Many drugs raise serum urate levels and predispose to gout attacks, especially diuretics.
Ultrasonography appears to be a useful adjunct in the diagnosis of acute and chronic gout.
Acute Gouty Arthritis
A single joint is involved in about 85% to 90% of first attacks, with the first metatarsophalangeal joint being the most commonly affected site. The initial attack is polyarticular in 3% to 14%. Acute gout is predominantly a disease of the lower extremities, but eventually, any joint of any extremity may be involved. Ninety percent of patients experience acute attacks in the great toe at some time during the course of their disease. Next in order of frequency are the insteps, ankles, heels, knees, wrists, fingers, and elbows. Acute attacks rarely affect the shoulders, hips, spine, sacroiliac joints, sternoclavicular joints, acromioclavicular joints, or temporomandibular joints.39,40 Acute gouty bursitis, tendinitis, or tenosynovitis can also occur.41,42 Urate deposition and subsequent gout appear to have a predilection for previously damaged joints such as in Heberden’s nodes of older women.43 The differential diagnosis is usually septic arthritis or other crystal-induced arthritis, but a broader differential should be considered in confusing cases (Table 95-1).
Table 95-1 Differential Diagnosis of Gout
| Acute Gouty Arthritis |
| Chronic Gouty Arthritis |
CPPD, calcium pyrophosphate disease.
Drugs may precipitate acute gout by either increasing or decreasing serum urate levels acutely. The occurrence of gout after the initiation of antihyperuricemic therapy is well established. In fact, the more potent the urate-lowering effect, the more likely there is to be an acute attack.44 Drug-induced gout secondary to increased serum urate levels occurs on occasion with diuretic therapy, intravenous heparin, and cyclosporine.45–47 Diuretic therapy in the elderly appears to be a particularly important precipitating factor for gouty arthritis. Other provocative factors include trauma, alcohol ingestion, surgery, dietary excess, hemorrhage, foreign protein therapy, infections, and radiographic contrast exposure.48,49 The risk of a patient with gout developing an attack during hospitalization is 20%.50
The definitive diagnosis of gout is best established by aspiration of the joint and identification of intracellular needle-shaped crystals that have negative birefringence with compensated polarized light microscopy. However, various alternatives have been proposed for a presumptive diagnosis. These include the triad of acute monoarticular arthritis, hyperuricemia, and a dramatic response to colchicine therapy,51 a set of criteria proposed by the American College of Rheumatology (Table 95-2) in 1977,52 and 10 “propositions” for diagnosis by a European League Against Rheumatism (EULAR) panel of experts in 2006 (Table 95-3).53 There are limitations to using any of these schemes. First, although the diagnosis of acute gouty arthritis can be strongly suggested by the typical presentation, not all inflammation of the great toe (podagra) in hyperuricemic patients is caused by gout.54 Second, some patients with gout are normouricemic at the time of an acute attack, a phenomenon related to alcohol use or a consequence of interleukin (IL)-6 generation by the acute inflammatory process.55–57 Third, diseases other than gout can occasionally improve with colchicine therapy; these include pseudogout, hydroxyapatite calcific tendinitis, sarcoid arthritis, erythema nodosum, serum sickness, rheumatoid arthritis, and familial Mediterranean fever.16 Finally, the simultaneous presence of both gout and septic arthritis can be confusing clinically, with the former masking the latter.58
Table 95-2 Criteria for the Classification of Acute Gouty Arthritis
Adapted from Wallace SL, Robinson H, Masi AT, et al: Preliminary criteria for the classification of acute arthritis of primary gout, Arthritis Rheum 20:895–900, 1977.
Table 95-3 Propositions and Strength of Recommendation (SOR): Order According to Topic (Clinical, Urate Crystals, Biochemical, Radiographic, and Risk Factors/Comorbidities)

The use of ultrasonagraphy as a means of diagnosing acute and chronic gout is gaining favor. The characteristic finding is a superficial, hyperechoic, irregular band on the surface of articular cartilage, the so-called “double contour sign” or “urate icing,” in one study seen in 92% of gouty joints and in no joints of patients with other types of arthritis.59,60 Also characteristic is nonhomogeneous tophaceous material surrounded by an anechoic rim. Further support for ultrasound comes from the demonstration of resolution of these findings in a small number of gout patients on urate-lowering therapy who maintained a serum urate less than or equal to 6 mg/dL for at least 7 months.61 Recent studies presented in abstract form have indicated excellent concordance between ultrasound readers in identifying changes, the presence of these findings even in those with asymptomatic hyperuricemia, and the superiority of ultrasound over conventional radiography in detecting gouty erosions.62–64 Ultrasound now appears to be a technology that will be an important ancillary approach to the diagnosis and treatment of gout. Magnetic resonance imaging is much more sensitive than even ultrasound at detecting gouty erosions in patients with gout and normal plain radiographs.65–67 Computed tomography (CT) scan is also sensitive for the detection of erosions and tophi, and three-dimensional CT may have utility in the quantitation of the size of tophi during clinical trials in gout.67
Intercritical Gout
The terms intercritical gout and interval gout have been applied to the periods between gouty attacks. Some patients never have a second attack. However, most patients suffer a second attack within 6 months to 2 years. In Gutman’s series,68 62% had recurrences within the first year, 16% in 1 to 2 years, 11% in 2 to 5 years, and 4% in 5 to 10 years; 7% had experienced no recurrence in 10 or more years. The frequency of gout attacks usually increases over time in untreated patients. Later attacks have a less explosive onset, are polyarticular, become more severe, last longer, and abate more slowly. Nevertheless, recovery is complete. Radiographic changes may develop during the intercritical period despite no sign of tophi on physical examination. These changes are more likely in patients with more severe hyperuricemia and more frequent acute attacks.50,69
The diagnosis of gout in a hyperuricemic patient with a history of acute attacks of monoarthritis may be difficult or inconclusive during the intercritical phase. Aspiration of an asymptomatic joint, however, can be a useful adjunct in the diagnosis of gout if urate crystals are demonstrated. Joint fluids obtained from gouty patients during the intercritical phase revealed monosodium urate crystals in 12.5% to 90% of joints.70 Such crystals in asymptomatic joints are often associated with mild synovial fluid leukocytosis, which suggests the potential to contribute to joint damage even in the intervals between attacks.
Chronic Gouty Arthritis
Eventually, the patient may enter a phase of chronic polyarticular gout with no pain-free intercritical periods. At this stage, gout may be easily confused with other types of arthritis or other conditions.71–73 The time from the initial attack to the beginning of chronic symptoms or visible tophaceous involvement is highly variable in studies of untreated patients. Hensch reported intervals ranging from 3 to 42 years, with an average of 11.6 years between the first attack and the development of chronic arthritis.74 Ten years after the first attack, about half the individuals were still free of obvious tophi and most of the remainder had only minimal deposits. Thereafter, the proportion of those with nontophaceous involvement slowly declined, to 28% after 20 years. Two percent of the patients had severe crippling disease some 20 years after the initial attack.
The rate of formation of tophaceous deposits correlates with both the degree and the duration of hyperuricemia. The principal determinant is the serum urate level.50 Gutman75 found the mean serum urate concentration to be 9.1 mg/dL in 722 patients without tophi, 10 to 12 mg/dL in 456 patients with minimal to moderate tophi, and greater than 11 mg/dL in 11 patients with extensive tophaceous involvement. The rate of tophus formation also increases with the severity of renal disease and the use of diuretics.45
Tophaceous gout is the consequence of the chronic inability to eliminate urate as rapidly as it is produced. As the urate pool expands, deposits of urate crystals appear in cartilage, synovial membranes, tendons, soft tissues, and elsewhere. Tophi are rarely present at the time of an initial attack of primary gout76,77; they are more likely to be present in gout secondary to myeloproliferative diseases, in juvenile gout-complicating glycogen storage diseases (GSDs), in Lesch-Nyhan syndrome, or after allograft transplantation in patients treated with cyclosporine.16,78
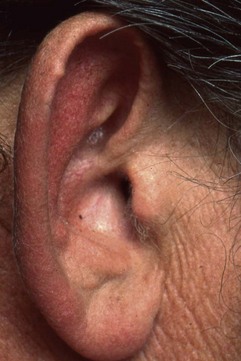
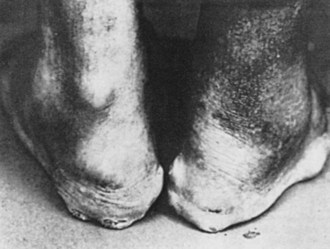
Tophi can occur in a variety of locations. Tophaceous deposits may produce irregular, asymmetric, moderately discrete tumescence of the fingers (Figure 95-1), hands, knees, or feet. Tophi also form along the ulnar surfaces of the forearm, as saccular distentions of the olecranon bursa (Figure 95-2), in the antihelix of the ear (Figure 95-3), or as fusiform enlargements of the Achilles tendon (Figure 95-4). The process of tophaceous deposition advances insidiously. Although the tophi themselves are relatively painless, acute inflammation can occur around them. Eventually, extensive destruction of the joints and large subcutaneous tophi may lead to grotesque deformities, particularly of the hands and feet, and to progressive crippling (Figure 95-5). The tense, shiny, thin skin overlying the tophus may ulcerate and extrude white, chalky, or pasty material composed of urate crystals. Secondary infection of tophi is rare.
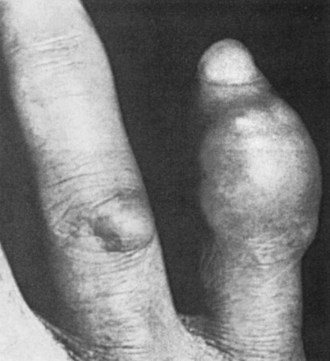
Figure 95-1 Tophus of the fifth digit, with a smaller tophus over the fourth proximal interphalangeal joint.
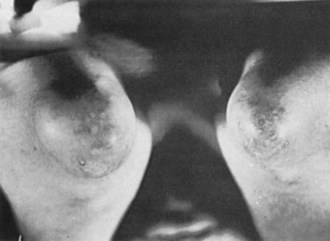
Figure 95-2 Saccular tophaceous enlargements of the oclecranon bursae, with small cutaneous deposits of urate.
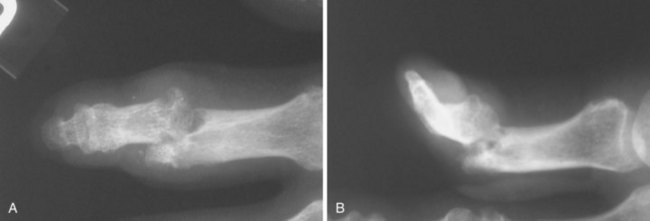
Typical radiographic changes, particularly erosions with sclerotic margins and overhanging edges of bone, occur with the development of tophi (Figure 95-6).79 These may be difficult to distinguish from erosions of other causes, but the presence of a thin, overhanging calcified edge is strong evidence of gout. Calcifications can be seen in some tophi, and bony ankylosis may rarely occur. Ultrasonography, magnetic resonance imaging, and computed tomography can demonstrate tophi, with the last providing the most specific images.59–6880
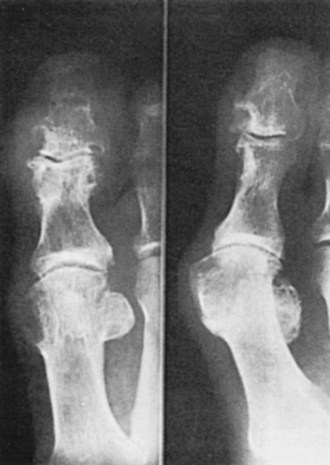
Tophi can produce a marked limitation of joint movement by involvement of the joint structure directly or of a tendon serving the joint. Any joint can be involved, although those of the lower extremity are affected primarily. Spinal joints do not escape urate deposition,73,81 but acute gouty spondylitis is unusual. Symptoms related to nerve or spinal cord compression by tophi have rarely been observed. Tophi rarely occur in myocardium, valves, cardiac conduction system, various parts of the eye, and larynx.82,83
Associated Conditions
Key Points
Renal insufficiency is frequently associated with hyperuricemia and gout.
A diagnosis of gout should prompt a search for the coexistence of these associated conditions.
The association of gout with obesity and overeating is well recognized.84 In 6000 subjects, hyperuricemia was found in only 3.4% of those with a relative weight at or below the 20th percentile, in 5.7% of those between the 21st and 79th percentiles, and in 11.4% of those at or above the 80th percentile.85
Hypertriglyceridemia has been reported in 75% to 80% of patients with gout,86 and hyperuricemia is found in more than 80% of patients with hypertriglyceridemia.16 However, studies have been unable to show a correlation between serum urate and cholesterol values or a unique lipid phenotype.87 Gouty patients who drink alcohol excessively have mean serum triglyceride levels that are higher than those of their obesity-matched controls and of non–alcohol-drinking gouty patients.88
Hyperuricemia has been reported in 2% to 50% of patients with diabetes mellitus, and gouty arthritis has been reported in less than 0.1% to 9%.89 Abnormal glucose tolerance tests have been noted in 7% to 74% of patients with gout, depending, in part, on the criteria used.90
Hyperuricemia has been reported in 22% to 38% of patients with untreated hypertension. This figure increases to 67% when diuretic therapy and renal disease are present.16 Hyperuricemia may be an indication of a potential risk for hypertension in adolescent males.91 Hypertension is present in one-fourth to one-half of patients with classic gout, but the presence of hypertension is unrelated to the duration of gout.84,85 Elevated serum urate concentrations are associated with increased tubular reabsorption of sodium.90 The serum urate concentration also correlates inversely with renal blood flow and urate clearance and correlates directly with both renovascular and total resistance. Therefore the association between hypertension and hyperuricemia may be related to the reduction of renal blood flow in hypertension. In addition, uric acid causes smooth muscle proliferation in vitro and vascular disease in animal models through a mechanism that involves complex intracellular signaling, mitogen-activated protein kinase activation, and platelet-derived growth factor expression.92,93
The association between hyperuricemia and the manifestations of atherosclerosis has led to speculation that hyperuricemia is a risk factor for coronary artery disease. Some studies show no clear associations among blood pressure, blood glucose, or serum cholesterol and serum urate concentration when adjustments are made for the effects of age, sex, and relative weight85,94–97; the serum urate concentrations of persons with coronary heart disease are not significantly different from the mean levels of the population.97,98 Other studies, however, maintain that hyperuricemia is an independent risk factor for coronary artery disease.99,100
The term metabolic syndrome has been applied to a cluster of abnormalities including resistance to insulin-stimulated glucose uptake, hyperinsulinemia, hypertension, and dyslipoproteinemia that are characterized by high levels of plasma triglycerides and high-density lipoprotein cholesterol. Hyperuricemia closely correlates with the degree of insulin resistance97–103 and therefore is a likely feature of metabolic syndrome. Metabolic syndrome has been associated with coronary artery disease, and hyperuricemia as a component of metabolic syndrome may explain the previously recognized association between coronary artery disease and hyperuricemia. A recent study concluded that the relationship between hyperuricemia and acute myocardial infarction is independent, but patients who experience gouty arthritis are at an increased risk for myocardial infarction. This association could not be explained by renal function, metabolic syndrome, diuretic use, or traditional cardiovascular risk factors.104
Alcohol consumption has long been associated with hyperuricemia and gout. In susceptible persons, alcohol use can precipitate acute gouty arthritis. An epidemiologic study in Saudi Arabia, where alcohol consumption is rare, revealed an 8.42% prevalence of hyperuricemia but no cases of gout among the study group.105 Both a decrease in the renal excretion of uric acid and an increase in uric acid production seem to be important factors in this association.106 Ethanol increases uric acid production by accelerating the turnover of ATP. Among alcoholic beverages, beer may have more potent effects on uric acid production because of its high guanosine content.14
There appears to be a significant increased prevalence of hypothyroidism among both female and male patients with gouty arthritis.107 Hyperuricemia may also be more prevalent in patients with hypothyroidism. Thyroid replacement therapy is associated with a decrease in serum urate concentration caused by an increased uric acid diuresis—a change not explained solely by a change in creatinine clearance.108 Although the cause of hyperuricemia and gout in patients with hypothyroidism is unknown, it is speculated that urate metabolism is mediated by thyroid-stimulating hormone receptors in extrathyroidal tissues including the kidney and that these modulate urate homeostasis.
Studies of acutely ill patients in intensive care units indicate that markedly increased serum urate concentrations, in the vicinity of 20 mg/dL, are associated with hypotensive events and a poor prognosis.109 This finding may be related to two factors. First, ischemic tissue may foster the degradation of ATP to purine end products, thereby enhancing the production of urate. The finding of increased plasma ATP degradation products associated with hyperuricemia and adult respiratory distress syndrome supports this possibility.110 Second, the conversion of hypoxanthine to uric acid by xanthine oxidase during ischemia produces oxidant radicals, which are themselves associated with tissue injury.111 It is possible that inhibition of xanthine oxidase with allopurinol may be a useful therapy in this setting.
Maternal serum urate concentrations normally decrease during pregnancy until the 24th week and then increase until 12 weeks after delivery.112 An increase in the serum urate level occurs in preeclampsia and toxemia of pregnancy, owing to a decrease in the renal clearance of urate.113 Perinatal mortality is markedly increased when maternal plasma urate levels are raised, usually in association with early-onset preeclampsia. The highest mortality rate is seen with serum urate concentrations higher than 6 mg/dL and diastolic blood pressures greater than 110 mm Hg. Labor itself is associated with an increased serum urate level, and it remains elevated for 1 to 2 days after delivery.
Gout is rarely seen in patients with rheumatoid arthritis, systemic lupus erythematosus, or ankylosing spondylitis.41,114–116 The basis for the decreased concurrence of these disorders is unclear, although the long-term use of nonsteroidal anti-inflammatory drugs (NSAIDs) or corticosteroids may mask the clinical features of gout in some of these patients.
Renal Disease
After gouty arthritis, renal problems appear to be the most frequent complication of hyperuricemia. Twenty percent to 40% of patients with gout have albuminuria, which is usually mild and often intermittent. Hyperuricemia alone may be implicated as the cause of chronic kidney disease only when the concentration of urate chronically exceeds 13 mg/dL in men or 10 mg/dL in women.117 Before the routine treatment of asymptomatic hypertension, renal failure accounted for 10% of the deaths in patients with gout. Whether moderate hyperuricemia has a direct harmful effect on renal function is unclear. Some evidence suggests that urate damages the kidneys and leads to hypertension.92,93
The term urate nephropathy is used to describe the deposition of urate crystals in the interstitium of the medulla and pyramids, with a surrounding giant cell reaction—a distinctive histologic finding characteristic of the gouty kidney (Figure 95-7). Factors such as coexistent hypertension, chronic lead exposure, ischemic heart disease, and primary pre-existing renal insufficiency probably play important roles in the pathogenesis of this pathology. Although urate nephropathy appears to exist as a distinct entity, it is not believed to be an important contributor to renal function in most gouty patients.16,118
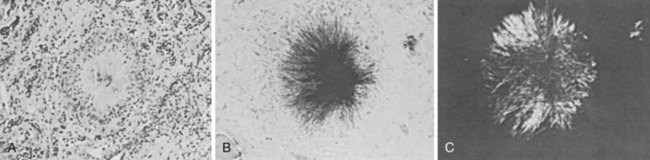
In contrast, uric acid nephropathy is the term used to describe acute renal failure resulting from the precipitation of large quantities of uric acid crystals in the collecting ducts and ureters. This complication most commonly occurs in patients with leukemia and lymphoma as a result of rapid malignant cell turnover, often during chemotherapy.119,120 This syndrome (also termed acute tumor lysis syndrome) has been more clearly defined as hyperuricemia, lactic acidosis, hyperkalemia, hyperphosphatemia, and hypocalcemia and is most commonly observed in patients with aggressive, rapidly proliferating tumors including lymphoproliferative disorders and metastatic medulloblastoma. Uric acid nephropathy is less commonly found with other neoplasms, after epileptic seizures, after vigorous exercise with heat stress, and after angiography and coronary artery bypass surgery.16
In the tumor lysis syndrome, the large amount of nucleic acid in nucleotides liberated with massive cytolysis is converted rapidly to uric acid. Typically, there is marked hyperuricemia, with a mean serum urate level of 20 mg/dL (range, 12 to 80 mg/dL). The pathogenesis of acute renal failure in uric acid nephropathy is related to the precipitation of uric acid in the distal tubules and collecting ducts, the sites of maximal acidification and concentration of urine. Oliguria, or even anuria, and azotemia may occur. There may be “gravel” or “sand” noted in the urine. The ratio of urinary uric acid to creatinine in these patients typically exceeds 1; in patients with most other causes of acute renal failure, the ratio is 0.4 ± 0.3.120
Nephrolithiasis occurs in 10% to 25% of patients with primary gout, a prevalence greater than that in the general population. The likelihood of stones in a given patient with gout increases with the serum urate concentration and with amounts of urinary uric acid excretion.121,122 It exceeds 50% with a serum urate value above 13 mg/dL or with urinary uric acid excretion rates in excess of 1100 mg every 24 hours.
Uric acid calculi account for approximately 10% of all stones in patients in the United States; elsewhere, rates range from as low as 5% up to 40% in Israel and Australia, respectively.16 Uric acid stones can occur in patients with no history of gouty arthritis, and only 20% in this group are hyperuricemic. Other renal stone disease is associated with hyperuricemia and gout. Gouty subjects also have an increased incidence of stones that contain calcium. In addition, about 30% of patients with recurrent calcium stone disease have either an increased urinary uric acid excretion rate or hyperuricemia. A causative link between uric acid and recurrent calcium oxalate stones is provided by reports of reduced stone frequency in patients treated with allopurinol.
Finally, the report of uric acid as the major constituent of a stone obtained from a patient with no apparent abnormalities of uric acid metabolism should suggest the possibility that the constituent is actually 2,8-dihydroxyadenine and that the patient has adenine phosphoribosyltransferase deficiency.123 This is because x-ray diffraction is required to distinguish uric acid from 2,8-dihydroxyadenine.
Familial juvenile hyperuricemic nephropathy (FJHN), sometimes called familial juvenile gouty nephropathy, was first described in 1960.124 This disorder is inherited as an autosomal dominant trait with a high degree of penetrance and is usually associated with gout. Renal disease typically develops in the second decade of life and progresses to end-stage renal failure by midlife.125–127 Histologic examination of kidney tissue reveals tubulointerstitial inflammation and splitting of thickened tubular basement membranes. The primary diagnostic criterion is a reduced fractional excretion of urate (defined as uric acid clearance factored by creatinine clearance × 100 equal to 5% or less; normal is 8% to 18%).128 The dramatically low fractional excretion of urate and the early onset of disease are conspicuous characteristics of FJHN and distinguish it from other autosomal dominant hyperuricemic disorders that usually appear later in life (Table 95-4). Genotype mapping has linked the gene for FJHN to chromosome 16p12-p11.126
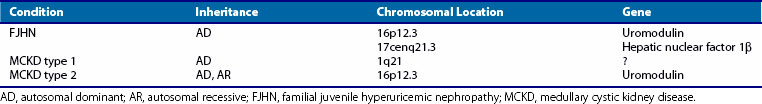
Autosomal dominant medullary cystic kidney disease (ADMCKD) is another hereditary nephropathy that usually includes gout among its constellation of symptoms. The onset of renal dysfunction occurs later than in those with FJHN. Renal histology reveals numerous corticomedullary and intramedullary cysts in the kidneys and increased medullary connective tissue. At least two loci appear to be responsible for ADMCKD. One, termed ADMCKD1, is located on chromosome 1; the other, ADMCKD2, is a 16p locus. ADMCKD2 and FJHN loci map to approximately the same region of chromosome 16p.129
The chromosome 16p12 locus that harbors the candidate interval for FJHN and ADMCKD2 contains six candidate genes, including the uromodulin gene (UMOD).130–132 UMOD encodes the Tamm-Horsfall protein, a glycosylphosphatidylinositol-anchored glycoprotein localized to the thick ascending limb of the loop of Henle. Amorphous deposits of uromodulin are present in the renal interstitium of patients with medullary cystic kidney disease.133 Four different mutations in exon 4 have been identified in the UMOD gene. Because mutations in the same gene are responsible for both FJHN and ADMCKD, the two entities appear to be allelic variants in UMOD that cause decreased urinary concentrations of Tamm-Horsfall protein, with resulting hyperuricemia and progressive renal failure.134
Lead Intoxication
Hyperuricemia and gout are well-recognized complications of chronic lead intoxication, with the prevalence of gout in patients with plumbism ranging between 6% and 50%.135 Although a renal defect is recognized, it has not been well defined.136,137 Some patients with primary gout have increased blood lead levels compared with age- and sex-matched controls, despite the absence of a history of overt lead exposure.138 This suggests that occult chronic lead intoxication may play a causative role in some cases of primary gout (up to 36% of some gout populations).135 In addition, patients with gout who have renal impairment seem to have an increased quantity of mobilized lead compared with gouty patients with normal renal function.139 These observations suggest an important role for lead in the pathogenesis of gouty nephropathy.
Cyclosporine-Induced Hyperuricemia and Gout
Cyclosporine interferes with the renal excretion of uric acid. Hyperuricemia and gout occur with increased frequency among transplant recipients treated with cyclosporine and are even more common when diuretics are used concomitantly.78,140 However, serum urate levels do not correlate directly with cyclosporine levels or with the degree of hypertension or renal insufficiency. The onset of gout may occur soon after transplantation, with a mean of about 17 months. Gouty attacks may be typical and monoarticular, or they may affect unusual sites such as the shoulder, hip, or sacroiliac joints. Polyarticular attacks and an accelerated course, with early development of tophi, may also be observed. Nephrolithiasis develops in about 3% of renal transplant patients. All calculi from azathioprine-treated patients are composed of calcium compounds, whereas 60% of the calculi from those treated with cyclosporine contain uric acid.141
Classification of Hyperuricemia and Gout
Hyperuricemia and gout may be classified as follows (Table 95-5):
Primary: These cases appear to be innate, neither secondary to an acquired disorder nor the result of a subordinate manifestation of an inborn error that leads initially to a major disease unlike gout. Some cases of primary gout have a genetic basis; others do not.
Secondary: These cases develop in the course of another disease or as a consequence of drug use.
Idiopathic: In these cases, a more precise classification cannot be assigned.
Table 95-5 Classification of Hyperuricemia and Gout
| Type | Metabolic Disturbance | Inheritance |
|---|---|---|
| Primary | ||
| Molecular defects undefined | ||
| Underexcretion (90% of primary gout) | Not established | Polygenic |
| Overproduction (10% of primary gout) | Not established | Polygenic |
| Associated with specific enzyme defects | ||
| PRPP synthetase variants; increased activity | Overproduction of PRPP and uric acid | X-linked |
| HPRT deficiency, partial | Overproduction of uric acid; increased purine biosynthesis de novo driven by surplus PRPP; Kelley-Seegmiller syndrome | X-linked |
| Secondary | ||
| Associated with Increased Purine Biosynthesis De Novo | ||
| HPRT deficiency, “virtually complete” | Overproduction of uric acid; increased purine biosynthesis de novo driven by surplus PRPP; Lesch-Nyhan syndrome | X-linked |
| Glucose-6-phosphatase deficiency or absence | Overproduction plus underexcretion of uric acid; glycogen storage disease type I (von Gierke’s disease) | Autosomal recessive |
| Fructose-1-phosphate aldolase deficiency | Overproduction plus underexcretion of uric acid | Autosomal recessive |
| Associated with Increased ATP Degradation | ||
| Associated with increased nucleic acid turnover | Overproduction of uric acid | Most not familial |
| Associated with decreased renal excretion of uric acid | Decreased filtration of uric acid, inhibited tubular secretion of uric acid, or enhanced tubular reabsorption of uric acid | Some autosomal dominant; some not familial; most unknown |
| Idiopathic | Unknown | |
ATP, adenosine triphosphate; HPRT, hypoxanthine phosphoribosyltransferase; PRPP, phosphoribosylpyrophosphate.
Further subdivisions within each major category are based on the identification of overproduction, underexcretion, or both, as responsible for the hyperuricemia. Evidence of overproduction of urate is provided by determination of the 24-hour urinary uric acid excretion. For adults ingesting a purine-free diet, a total excretion of up to 600 mg/day is considered within the normal range.142 For patients on regular diets, a value in excess of 1000 mg/day is clearly abnormal and an indication of overproduction, and values between 800 and 1000 mg/day are considered borderline. It has been suggested that overproduction of uric acid can be assessed simply by determining the ratio of uric acid to creatinine in the urine or the Curate/Ccreatinine ratio. However, comparison of these two ratios with the 24-hour urinary uric acid excretion reveals a poor correlation in most patients.143,144 Exceptions include patients with specific enzymatic deficiencies or with rapid cell lysis during chemotherapy for leukemia or lymphoma.
Primary Gout
Renal mechanisms are responsible for the hyperuricemia in most cases of gout. Genetic factors exert an important control in the renal clearance of urate.145 A careful comparison of uric acid clearances and excretion rates over a wide but comparable range of filtered loads of urate indicates that most gouty subjects have a lower ratio of urate-to-inulin clearance (Curate/Cinulin ratio) than do nongouty subjects.142,145,146 The excretion rates and the capacity of the excretory mechanism for uric acid are the same for gouty subjects and nongouty individuals (see Figure 94-8). The excretion curve, however, is shifted. Gouty subjects require serum urate values 2 or 3 mg/dL higher than those of controls to achieve equivalent uric acid excretion rates. Theoretically, the shift in the excretion curve in gouty subjects may result from reduced filtration of urate, enhanced reabsorption, or decreased secretion. Patients classified as exhibiting an overproduction of uric acid represent less than 10% of the gouty population.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


