69 Etiology and Pathogenesis of Rheumatoid Arthritis
Adaptive and innate immune responses in the synovium have been implicated in the pathogenesis of RA.

 Video available on the Expert Consult Premium Edition website.
Video available on the Expert Consult Premium Edition website.
Etiology and Pathogenesis of Rheumatoid Arthritis: Roles of Innate and Adaptive Immunity
The etiology and pathogenesis of RA are complex and multifaceted. A variety of predetermined (genes) and stochastic (random events and environment) factors contribute to susceptibility and pathogenesis. As an introduction, a summary of how these mechanisms interact to create and perpetuate RA is shown in Figure 69-1. Individual mechanisms are discussed in greater detail throughout the chapter.
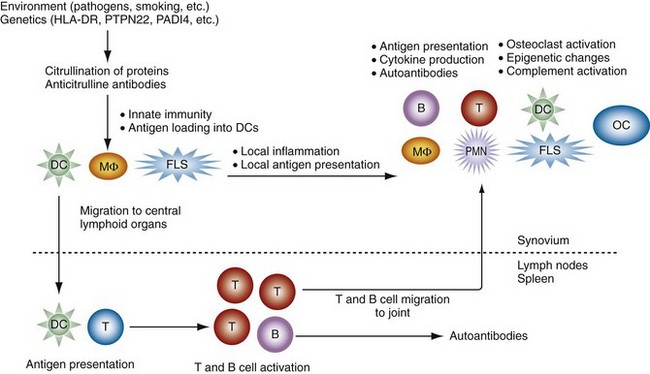
The initiation of RA probably begins years before the onset of clinical symptoms. This process involves certain specific genes that can help break tolerance and lead to autoreactivity. It is likely that the earliest phases are marked by repeated activation of innate immunity (see Figure 69-1).1 Cigarette smoke, bacterial products, viral components, and other environmental stimuli can contribute to these responses. This process probably occurs often in normal individuals but is self-limited. In individuals, a predetermined propensity for immune hyper-reactivity or autoreactivity might lead to a different outcome. The genome of these individuals might encode for a variety of genes implicated in RA including class II major histocompatibilty complex (MHC) genes, protein tyrosine phosphatase-22 (PTPN22), cytokine promoter polymorphisms, signal transduction gene polymorphisms, population-specific genes (e.g., PADI4 in Japanese or Koreans), and other undefined genes. Abnormal T cell selection could also contribute by allowing autoreactive T cells to escape deletion. The environmental stresses can lead to post-transcriptional modification of proteins, especially citrullination of arginine residues, in mucosal surfaces or the synovium. Although this commonly occurs without sequelae in normal individuals, people with a propensity for RA can develop antibodies against these modified proteins with production of rheumatoid factors (RFs) and anticitrullinated protein antibodies (ACPAs).
Etiology of Rheumatoid Arthritis
Key Points
Genes play a key role in susceptibility to RA, as well as disease severity.
Although the immunogenetics is, at best, incompletely understood, one of the best-studied and perhaps most influential genetic risk factor is the class II MHC haplotype of an individual. PTPN22 and PADI4 increase risk in some racial and ethnic groups, but not all. Genome-wide screens have implicated at least 35 genes, many of which are involved with immune function. However, most have a relatively modest contribution and the susceptibility polymorphism confers only a 1.1- or 1.2-fold increase. Combinations of genes can clearly interact with one another, and a 45-fold increase in risk is conferred by a combination of HLA-DR, PTPN22, and the TRAF1-C5.1 This combination, however, is found in less than 1% of individuals with RA. The RA-associated alleles identified to date contribute approximately 40% of total genetic susceptibility. Additional progress in understanding the role of genes in RA including rare variants that might be more important than some common polymorphisms will require sophisticated bioinformatics to clarify how individual alleles contribute to susceptibility, severity, and response to targeted therapies.
Role of HLA-DR in the Susceptibility to and Severity of Rheumatoid Arthritis
The susceptibility to RA is associated with the third hypervariable region of DRβ-chains, from amino acids 70 through 74. The epitope is glutamine-leucine-arginine-alanine-alanine (QKRAA), a sequence found in DR4 and DR14, in addition to some DR1β-chains. The “susceptibility epitope” (SE) on DR4β-chains with the greatest association with RA are DRB*0401, DRB*0404, DRB*0101, and DRB*1402 (Table 69-1). Up to 96% of patients with RA exhibit the appropriate HLA-DR locus in some populations.2 In certain ethnic and racial groups, however, the association with QKRAA is not as prominent or is not associated. The QKRAA epitope might also predict the severity of established RA, with a greater prevalence of extra-articular disease and erosions in patients with two copies. Other HLA genes such as DRB*1301 contain the DERAA sequence and are associated with decreased susceptibility to RA.3
Table 69-1 Nomenclature for HLA-DR Alleles and Associations with Rheumatoid Arthritis
| Old Nomenclature (HLA-DRB1 Alleles) | Current Nomenclature | Association with Rheumatoid Arthritis |
|---|---|---|
| HLA-DR1 | 0101 | + |
| HLA-DR4 Dw4 | 0401 | + |
| HLA-DR4 Dw14 | 0404/0408 | + |
| HLA-DRw14 Dw16 | 1402 | + |
| HLA-DR4 Dw10 | 0402 | − |
| HLA-DR2 | 1501, 1502, 1601, 1602 | − |
| HLA-DR3 | 0301, 0302 | − |
| HLA-DR5 | 1101-1104, 1201, 1202 | − |
| HLA-DR7 | 0701, 0702 | − |
| HLA-DRw8 | 0801, 0803 | − |
| HLA-DR9 | 0901 | − |
| HLA-DRw10 | 1001 | − |
| HLA-DRw13 | 1301-1304 | 1301 associated with protection |
| HLA-DRw14 Dw9 | 1401 | − |
Modified from Weyand CM, Hicok KC, Conn DL, Goronzy JJ: The influence of HLA-DRB1 genes on disease severity in rheumatoid arthritis, Ann Intern Med 117:801, 1992.
One intriguing possibility that could account for some patients who do not fit within this paradigm is microchimerism.4 Maternal cells expressing the SE can survive and persist in the circulation throughout adulthood. These noninherited maternal antigens (NIMAs) could then confer increased risk of disease in the children of SE-expressing women.
The shared epitope might not be an independent risk factor for RA but instead a marker for immunoreactivity and anticitrullinated protein antibodies (ACPAs).5 In a large series of patients with early undifferentiated inflammatory arthritis, one-third of patients met criteria for RA within 1 year. Progress to RA occurred regardless of HLA-DR genotype if patients were anticitrullinated protein (anti-CP) positive. When patients were stratified according to ACPA, the shared epitope did not make an additional contribution to progression from undifferentiated arthritis to RA. The shared epitope probably contributes to immune hyper-reactivity, but ACPAs are more closely associated with RA. In other studies, however, the presence of the shared epitope and ACPAs together is associated with even greater disease severity.
Additional Polymorphisms: Cytokines, Citrullinating Enzymes, PTPN22, and Others
The genetic influence on RA has also led to studies evaluating non-MHC genes. Single nucleotide polymorphisms (SNPs) in promoter regions, coding regions, or areas with no known function have been extensively investigated in RA with a variety of methods including genome-wide association studies. Table 69-2 shows some of the SNPs and microsatellites that have been associated with RA. The relative contribution for most is modest, and variations in technique, stage of disease, and patient populations result in some disagreement among various reports.
Table 69-2 Key Genetic Associations in Rheumatoid Arthritis
| Gene | Odds Ratio for Risk Alleles | Comment |
|---|---|---|
| HLA-DR | 4-5 fold | |
| PTPN22 | ≈2 fold | Not in Asian populations |
| PADI4 | ≈2 fold | Primarily in Asian populations |
| TRAF1-C5 | >1.2 fold; <2 fold | |
| STAT4 | >1.2 fold; <2 fold | |
| TNFAIP3 | >1.2 fold; <2 fold | |
| IL2/21 | >1.2 fold; <2 fold |
Other genes with odds ratio >1.0 and <1.2: CTLA4, CD40, CCL21, CD244, IL2Rb, TNFRSF14, PRKCQ, PIP4K2C, IL2RA, AFF3, REL, BLK, TAGAP, CD28, TRAF6, PTPRC, FCGR2A, PRDM1, CD2-CD58, IRF5, CCR6, CCL21, IL6ST, RBPJ.
Given the importance of cytokines in RA (see following), it is not surprising that many studies have focused on these genes. The most intriguing evidence relates to tumor necrosis factor (TNF). This proinflammatory factor is a major cytokine in the pathogenesis of RA, and the TNF genes are located in the MHC locus on chromosome 6 in humans. Several polymorphisms of the TNF promoter including two at positions −238 and −308 can alter gene transcription. Associations among the TNF polymorphisms and RA susceptibility and radiographic progression have been reported, although there is not uniform agreement. In addition, certain polymorphisms in cytokines, especially TNF or Fc receptors, have been associated with differential response to therapy. For instance, substitution of a T for a C at position −857 in the TNF promoter might confer greater responsiveness to TNF inhibitors.6
Among the many noncytokine and non-MHC genetic linkages described for RA, the ones associated with peptidyl arginase deiminase (PADI) and PTPN22 have the strongest effect on susceptibility. The PADI genes are responsible for the post-translational modification of arginine to citrulline. Four isoforms have been identified, known as PADI1 through PADI4. In light of the striking associations of RA with ACPAs, several groups have investigated potential associations with these genes. The most promising is an extended haplotype in the PADI4 gene that can lead to increased levels of PADI4 protein due to enhanced messenger RNA (mRNA) stability.7 In a Japanese cohort, a twofold increase in risk of RA was observed with PADI4 SNPs. Confirmatory reports have been mixed because the association has been confirmed in other Asian populations but not in Western Europe. These studies suggest that the contribution of PADI4 to RA might be restricted, depending on the overall genetic background of the patient population.
Protein tyrosine phosphatase-22 (PTPN22) associations have been discovered in large-scale screening efforts to identify SNP associations in RA.8 Using 12,000 SNPs in the initial screens, a novel association was discovered at position 1858 in the PTPN22 gene that, like PADI4, conferred a twofold increase in risk. The allele containing thymidine leading to an amino acid substitution (R620W) was present in 8.5% of controls but was found in nearly 15% in patients with seropositive RA. Subsequent studies have demonstrated a similar association with systemic lupus erythematosus (SLE), type 1 diabetes, and several other autoimmune diseases. PTPN22 is a phosphatase that regulates the phosphorylation status of several kinases important to T cell activation including Lck and ZAP70. The R620W allele surprisingly results in a gain of function that alters the threshold for T cell receptor (TCR) signaling. Because the PTPN22 allele is rare in Japan, it is another gene (e.g., PADI4) where susceptibility is specific for particular ethnic or racial populations.
The list of genes associated with RA consistently involves immune regulation.9 Cytokine polymorphisms such as for TNF and the IL-1 inhibitor, IL-1Ra are not surprising. Genes that regulate adaptive immune responses in T cells such as PTPN22 and the co-stimulation receptor CTLA have also been associated with RA. Other genes associated with B cell function and/or antigen presentation such as BTLA (B- and T-lymphocyte attenuator), Fc receptors, and CD40 are also implicated. Polymorphisms have also been identified in signal transduction pathways that regulate immune function such as TRAF1-C5 and STAT4. The consistent thread in this analysis is that most gene associations for RA cluster to innate immunity, adaptive immunity, and inflammation. Aside from providing insight into the mechanisms of disease, they could also potentially contribute to responses to targeted therapies.
Interactions between Genes and Environment
A number of environmental factors clearly contribute to RA susceptibility, although no specific exposure has been identified as the pivotal agent. Smoking is the best defined environmental risk factor for seropositive RA. The reason for its influence on the development of synovitis is not fully defined but could involve the activation of innate immunity and PADI in the airway. Citrullinated proteins have been detected in bronchoalveolar lavage samples of smokers, and this could provide a stimulus for generation of ACPAs in susceptible individuals.10 Repeated activation of innate immunity, especially in an individual with underlying genetically determined autoreactivity, could potentially contribute to autoreactivity and the initiation of synovitis. Other environmental factors such as oral contraceptives appear to modestly protect from RA, perhaps due to changes in the hormone milieu.11
The interaction between HLA-DR and tobacco exposure is perhaps the best example of how genes and the environment conspire to enhance risk. Although smoking and the SE alone modestly increase the likelihood of developing RA, the combination is synergistic.12 An individual with a history of cigarette smoking and two copies of the SE increases the odds of developing RA by up to 40-fold. The mechanism of the interaction is not known, but it could potentially relate to the increase in protein citrullination in smokers and increased ability of SE-containing HLA-DR molecules to bind some citrullinated proteins. The extent of smoking is also predictive, with the greatest risk seen with at least 20 pack-years. The risk declines slowly with cessation of smoking, taking more than a decade to begin approaching nonsmokers.13 Alcohol consumption can decrease this risk, and exposure to other inhaled irritants like silica dust increases risk, demonstrating the complexity of environment and human behavior on understanding disease susceptibility.
Gender
RA is one of many chronic autoimmune diseases that predominates in women. The ratio of female-to-male patients is 2 : 1 to 3 : 1, which in not as high as Hashimoto’s thyroiditis (25 : 1 to 50 : 1) or SLE (9 : 1). The gender effect is often observed in some animal models of autoimmunity such as the NZB/NZW model of SLE, in which female mice have more severe disease. Estrogens are one obvious explanation, and some data support the concept that these hormones modulate immune function.14 For example, autoantibody-producing B cells exposed to estradiol are more resistant to apoptosis, suggesting that autoreactive B cell clones might escape tolerance. The effect on T lymphocytes is harder to reconcile with the female preponderance in RA because estrogens tend to bias T cell differentiation toward the Th2 phenotype. The cytokines produced by this subset such as IL-4 and IL-13 are usually considered anti-inflammatory in animal models of arthritis and are present in only limited amounts in the RA synovium. Estrogen receptors are expressed on fibroblast-like synoviocytes (FLS) and increase production of metalloproteinases. In macrophage cell lines, estrogen can enhance production of TNF. Nulliparity has also been suggested as a risk factor in early studies, but more recent reports do not support this notion. Thus the effects of estrogens are complex, and the specific mechanisms responsible for the female preponderance of RA are not fully understood.
Pregnancy is often associated with remission of the disease in the last trimester. More than three quarters of pregnant patients with RA improve in the first or second trimester, but 90% of these experience a flare of disease associated with a rise in RF titers in the weeks or months after delivery. The mechanism of protection is not defined but might be due to the expression of suppressive cytokines such as IL-10 during pregnancy, production of α-fetoprotein, or alterations in cell-mediated immunity. One intriguing finding is that fetal DNA levels in the maternal peripheral blood correlate with the propensity for improved symptoms in pregnant RA patients. It is not certain whether the DNA itself contributes or whether it is a marker for increased leakage of fetal cells into the maternal circulation.15 Immune responses directed against paternal HLA antigens can occur and lead to the production of alloantibodies in the maternal circulation. Maternal-fetal disparity in human leukocyte antigen (HLA) class II phenotypes can correlate with pregnancy-induced remission. More than three-fourths of pregnant women with maternal-fetal disparity of HLA-DRB1, DQA, and DQB haplotypes have significant improvement, whereas disparity is only observed in one-fourth of women whose pregnancy is characterized by continuous active arthritis.16 Therefore suppression of maternal immune responses to paternal HLA haplotypes might be protective. This question remains unsettled because another study failed to find a correlation between the HLA disparity and clinical improvement during pregnancy.17
Epigenetics
Most information on epigenetics in RA comes from studies of RA synovium or cultured synoviocytes. Some evidence of imprinting is available in the latter, with evidence of global DNA hypomethylation.18 Only low levels of a key DNA methylase, Dnmt1 are expressed in RA synovium, suggesting a molecular basis for this observation. Because this enzyme is carried by gametes, the methylation pattern can be transgenerationally maintained. One particular CpG site in the IL-6 promoter of peripheral blood mononuclear cells has decreased methylation, and this is associated with higher levels of IL-6 production.19 Dietary influences such as ingestion of methyl donors such as folate, or even exposure to methyl donors in utero, can profoundly alter DNA methylation and adaptive immune functions and affect susceptibility to autoimmune disease.
The histone deacetylase HDAC1 is overexpressed in RA FLS. When this gene is suppressed, synoviocyte proliferation decreases and expression of tumor suppressor proteins such as p53 increases. HDAC inhibitors are also effective in collagen-induced arthritis, markedly delaying the onset of disease and decreasing bone erosions. Finally, analysis of synovioyctes shows that some individual microRNAs such as microRNA-124a are decreased in RA compared with osteoarthritis (OA) cells. This particular microRNA can suppress cell cycling and chemokine genes. Increasing microRNA-124a levels in RA synoviocytes decreased the production of the chemokine MCP-1.20 Forced expression of microRNA-203 increases metalloproteinase and IL-6 expression by synoviocytes as well.21
Changing Epidemiology of Rheumatoid Arthritis
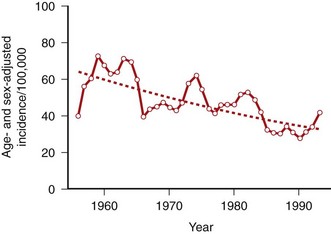
Equally intriguing, the severity and incidence of RA appeared to decrease in the late twentieth century (Figure 69-2).22 In certain well-defined populations including Native Americans, the incidence of RA has gradually declined by as much as 50% over the past half of the twentieth century. Changes in hygiene and other lifestyle modifications related to industrialization might contribute, and an infectious agent might be less prevalent secondary to these societal changes, as with many other infectious diseases. Recent data from 1995 to 2007 suggest that the incidence might be rising again in women, but not in men. Dissecting the environmental exposures will be key to understanding how the susceptibility to disease varies over time.23
Pathogenic Mechanisms in Rheumatoid Arthritis
Key Points
A single specific “RA pathogen” is unlikely.
Considerable effort has been expended to assess the role of infectious agents in RA (Table 69-3). A potential pathogen could initiate disease through a variety of mechanisms including direct infection of the synovium, activation of innate immunity by pattern-recognition receptors that bind to components of the agent, or through molecular mimicry that induces an autoreactive adaptive immune response.
Table 69-3 Etiology of Rheumatoid Arthritis: Possible Infectious Causes
| Infectious Agent | Potential Pathogenic Mechanisms |
|---|---|
| Mycoplasma | Direct synovial infection; superantigens |
| Parvovirus B19 | Direct synovial infection |
| Retroviruses | Direct synovial infection |
| Enteric bacteria | Molecular mimicry (QKRAA, e.g., in bacterial heat shock proteins) |
| Mycobacterium | Molecular mimicry (proteoglycans, QKRAA), immunostimulatory DNA (Toll-like receptor 9 activation) |
| Epstein-Barr virus | Molecular mimicry (QKRAA in gp110) |
| Bacterial cell walls | Toll-like receptor 2 activation |
Infectious Agents: Direct Infection and Innate Immune Responses
Toll-like Receptors and the Inflammasome in the Joint
The role of innate immunity in RA led to the notion that repeated engagement of TLRs in the synovium could help initiate disease. This hypothesis could explain why specific pathogens have been difficult to identify in the joint. In contrast, a genetically susceptible individual could potentially break tolerance if the TLRs are repeatedly engaged and permit autoimmune responses against articular antigens. Several animal models of disease require TLR ligands for initiations such as TLR9 in adjuvant arthritis. TLR2 is required for streptococcal cell wall arthritis, and the chronic T cell–dependent phase of that model requires TLR4. Mice lacking TLR4−/− have significantly less joint damage induced by IL-1 overexpression, even though synovial inflammation is still robust.24 These data suggest that endogenous TLR ligands play a key role in matrix regulation independent of inflammatory responses.
Bacteria, Mycobacteria, Mycoplasma, and Their Components
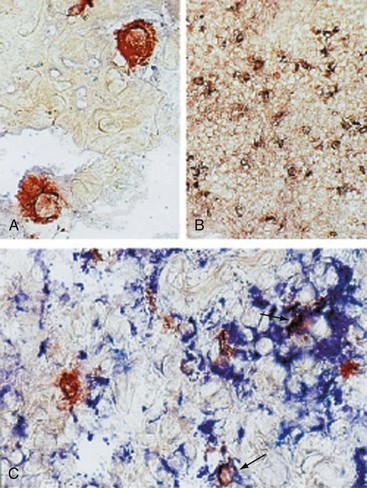
In addition to prokaryotic DNA, bacterial peptidoglycans have been detected in RA synovial tissue (Figure 69-3). Antigen-presenting cells containing these products express TLRs and produce proinflammatory cytokines such as TNF. It is not known whether the peptidoglycans activate cells in situ or whether phagocytic cells from other sites or the blood engage the molecules and then migrate to the joint. In either case, it is not difficult to imagine how they can contribute to synovial inflammation.
Several animal models of arthritis are dependent on TLR2, TLR3, TLR4, or TLR9. For instance, rodents injected with streptococcal cell walls (TLR2 ligand) develop severe polyarticular arthritis. The initial phase of disease resolves and is then followed by a chronic T cell–dependent phase that resembles RA. The arthritogenicity of complete Freund’s adjuvant in the rat adjuvant arthritis model is dependent on mycobacterial DNA that binds to TLR9 and activates an adaptive immunity. Endogenous TLR4 ligands such as heat shock proteins and fibrinogen also play a role in immune complex models such as passive K/BxN arthritis.25
Parvovirus
The mechanisms of B19-induced synovitis, when it does occur, could be related to alterations in the function of FLS.26 In a cell-culture model of synoviocyte invasion into cartilage, infection with the parvovirus significantly increased the migration of cells into the matrix. Mice that are transgenic for the B19 protein NS1 were more susceptible to collagen-induced arthritis and developed high titers of anti–type II collagen antibodies. These data suggest that the B19 genome might not cause arthritis but can enhance an arthritogenic response to other environmental stimuli.
Autoimmunity
Key Points
Evidence of autoimmunity can be present in RA many years before the onset of clinical arthritis.
Autoantibodies such as RFs and anticitrullinated protein antibodies are commonly associated with RA.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


