94 Etiology and Pathogenesis of Hyperuricemia and Gout
 Video available on the Expert Consult Premium Edition website.
Video available on the Expert Consult Premium Edition website.
 The ancient disease gout has a complex pathogenesis, and its modern relevance is underscored by a rise in prevalence by as much as fourfold in the past half century. Indeed, gout is now the most common inflammatory arthritis in the United States.1 Gout is a disease of both metabolism and inflammation/immunity. Gout pathogenesis requires the intersection of two distinct processes: (1) the intrinsic formation of uric acid, in the form of urate, at levels sufficient to drive the precipitation of urate into crystallized forms and (2) an inflammatory response to the crystals so formed. How these processes occur and under what circumstances they cross from adaptive to pathologic responses are the subjects of this chapter.
The ancient disease gout has a complex pathogenesis, and its modern relevance is underscored by a rise in prevalence by as much as fourfold in the past half century. Indeed, gout is now the most common inflammatory arthritis in the United States.1 Gout is a disease of both metabolism and inflammation/immunity. Gout pathogenesis requires the intersection of two distinct processes: (1) the intrinsic formation of uric acid, in the form of urate, at levels sufficient to drive the precipitation of urate into crystallized forms and (2) an inflammatory response to the crystals so formed. How these processes occur and under what circumstances they cross from adaptive to pathologic responses are the subjects of this chapter.
Evolutionary Considerations
Uric Acid as a Danger Signal
Uric acid is a breakdown product of purine metabolism. As such, it represents a metabolic waste molecule that might, in theory, be nothing more than a nuisance requiring excretion. However, studies by Shi and colleagues2 and others suggest that evolution has co-opted this waste-generating process to play an important and perhaps critical role in organismal immunity. It had long been appreciated that the lysates of damaged mammalian cells can serve effectively as adjuvants—that is, can promote immune responses to injected antigens. Recently Shi and colleagues2 used classical biochemical techniques to demonstrate that the major endogenous adjuvant found in damaged cells was uric acid. These investigators further demonstrated that uric acid had the capacity to promote T cell activation in response to antigen and that aggressive urate-lowering treatment could abrogate murine immune responses. Thus uric acid may serve as a danger signal to promote immune responses. As first proposed by Matzinger, a danger signal is an intrinsically produced molecule, typically issued by an altered or damaged cell in order to alert the immune system to the need for an immunologic response.3 Viewed from this perspective, the production of uric acid in a virally infected cell, for example, might serve as an upstream “second signal” to promote antigen presentation by a professional antigen-presenting cells such as dendritic cells, macrophages, or B cells. Indeed, although damaged or dying cells tend to have limited ability to manufacture proteins, their output of uric acid characteristically increases during cellular breakdown. The uric acid danger signal might also play an important role in tumor immunity, and at least one mouse model suggests that modulation of uric acid levels may directly affect immune tumor rejection.4 Although these observations require more study, they are consistent with a paradigm in which urate production at the local cellular level may play an important role in immunity and homeostasis.
Uric Acid and Human Evolution
Most mammals have serum urate levels in a range roughly between 0.5 and 2 mg/dL. In contrast, humans and other primates, including some New World monkeys, typically demonstrate serum urate levels between 4 and 6 mg/dL. The genetic and biochemical basis for these increases is well appreciated. During the Miocene era (10 to 25 million years ago), mutations in various primate and some monkey species resulted in inactivation of the uricase gene, which codes for the enzyme that degrades uric acid to allantoic acid. Genetic studies indicate that the uricase gene experienced nonsense mutations during that period, not once but multiple times across multiple hominoid lineages (Figure 94-1).5,6 The occurrence of multiple independent loss-of-function mutations has led some biologists to hypothesize that increases in urate generation may have conferred a survival benefit for these particular species. Several compelling, and not necessarily mutually exclusive, hypotheses have been proposed.
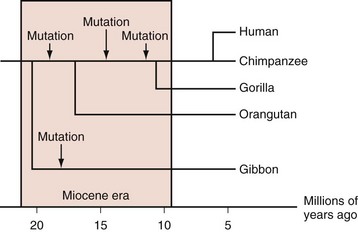
Ames and colleagues7 noted the fact that these same primate species had also been subject to a unique deletion of the gene permitting organisms to produce ascorbic acid, an event that apparently occurred in the Eocene era, some 10 to 20 million years before the uricase deletions. In mammals that do produce ascorbic acid, this molecule serves as the pre-eminent antioxidant in the body. Thus the loss of ascorbate production may have been an evolutionary liability, for which increases in urate provided compensation, particularly as a protectant against aging and cancer.7 Other authors have suggested that the effects of urate may have been particularly important in the central nervous system and that hyperuricemia provided an evolutionary advantage by promoting hominoid intellectual function, either through its antioxidant effects or via activation of neurostimulatory adenosine receptors (in a manner similar to that of caffeine).8 Although the antioxidant theory would appear compelling, critics have pointed out that (1) the production of urate itself generates oxidant molecules, diminishing any possible urate benefit; (2) intracellular urate may have pro-oxidant effects9; and (3) the human/primate capacity for antioxidation may be large even in the absence of soluble antioxidants; for example, human red cell membranes have extensive antioxidant capacities.10
Other investigators have examined the specific evolutionary pressures exerted on primate species during the Miocene era, in an attempt to understand the potential advantages of urate elevations. Johnson and colleagues12 pointed out that the Miocene era was an important era in hominoid evolution and that the hominoid diet during that time appears to have been mainly vegetarian and extremely low in salt. They suggest that hominoids during that period may have experienced a “hypotensive crisis,” particularly in the face of the transition to upright walking. They further postulate that an elevation in serum uric acid levels provided a mechanism for restoring normotension, primarily through urate-induced renovascular injury.11,12 To model this hypothesis, these investigators exposed rats to a low-salt diet, resulting in hypotension. When treated with the uricase inhibitor oxonic acid (to mimic the primate uricase loss), the rats’ urate levels rose and their blood pressures normalized. The effects of uricase inhibition on blood pressure could be reversed through the use of the urate-lowering agent allopurinol. These observations imply that what may once have been a homeostatic adaptation could now contribute to essential hypertension in today’s salt-rich era. In support of the latter hypothesis, Feig and colleagues13 identified adolescents with premature essential hypertension and hyperuricemia and treated them with the urate-lowering agent allopurinol. The result was normalization in blood pressure that reversed after allopurinol discontinuation.
Uric Acid Production and Excretion: Normal Levels and Hyperuricemia
Uric acid is a breakdown product of purines, and uric acid generation therefore depends directly on both intrinsic purine production and purine intake. In humans, uric acid is an end-product metabolite; consequently the depletion of uric acid depends directly on its excretion. The balance between uric acid production and excretion determines the serum urate level. Most individuals maintain a relatively stable uric acid level between 4 and 6.8 mg/dL and a total body uric acid pool of approximately 1000 mg.14 However, it is increasingly appreciated that individuals with high serum uric acid levels may deposit uric acid either occultly or in the form of appreciable masses (tophi), with the consequence that the total body urate pool may be significantly higher than in nonhyperuricemics.15 Such occult deposition of uric acid (total body urate burden) may have implications for treatment because they may form a “buffering reservoir” of urate that resists initial treatment with urate-lowering agents.
Urate Production: Purine Metabolism and Intake
Purine Biosynthesis
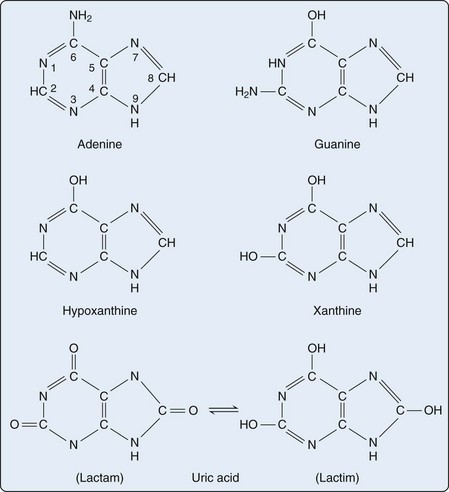
Purines are heterocyclic aromatic compounds, consisting of conjoined pyrimidine and imidazole rings (Figure 94-2). In mammals, the most common expression of purines is found in the form of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) (containing the purines adenine and guanine), as well as single-molecule nucleotides (adenosine triphosphate [ATP], adenosine diphosphate [ADP], adenosine monophosphate [AMP], cyclic AMP, and to a lesser extent, guanosine triphosphate [GTP] and cyclic glucose monophosphate [GMP]). Purines are also critical elements of the energy metabolism molecules NADH, NADPH, and coenzyme Q. Purines may also serve as direct neurotransmitters; for example, adenosine may interact with receptors to modulate cardiovascular and central nervous system function.16
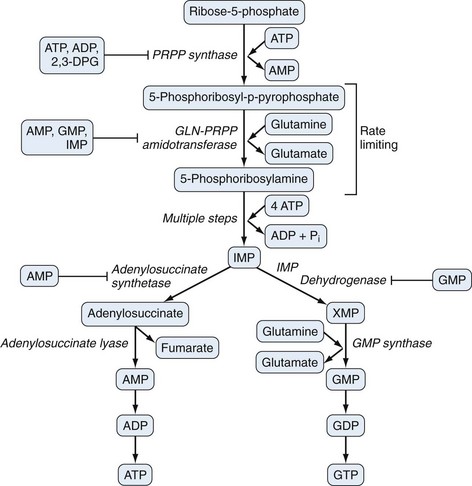
Purine biosynthesis is initiated on a core of ribose-5-phosphate (Figures 94-3 and 94-4). The enzyme phosphoribosyl pyrophosphate (PRPP) synthase catalyzes the addition of a pyrophosphate moiety to form the adduct PRPP. This reaction is thought to be rate limiting. Subsequently, the enzyme glutamine-PRPP amidotransferase catalyzes the interaction of PRPP with glutamine to form 5-phosphoribosyl amine, the commitment step in purine biosynthesis. Glutamine-PRPP amidotransferase and PRPP synthase are both subject to feedback inhibition by IMP, AMP, and GMP, providing a mechanism to slow purine biosynthesis in the setting of purine sufficiency. 5-phosphoribosyl amine next forms the backbone for a series of molecular additions, ending in the formation of the purine inosine monophosphate (IMP). IMP is converted into either adenosine monophosphate (AMP) or guanosine monophosphate (GMP), which can then be phosphorylated into higher-energy compounds. Collectively, the process of purine biosynthesis is highly energy dependent, requiring the consumption of multiple molecules of ATP. Thus purine biosynthesis not only directly increases the substrate load for urate generation but also increases the turnover of already-formed purines that contribute to increased urate levels.17
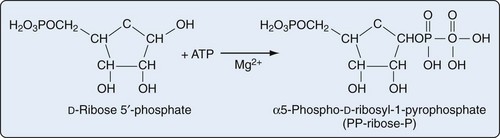
Urate Formation and Purine Salvage
Purines generated by the previously described mechanisms are susceptible to enzymatic catabolism, presumably to maintain purine homeostasis (Figure 94-5). Purines susceptible to degradation include the monophosphate nucleotides GMP and IMP. These molecules are converted by nucleotidases to their purine base forms, guanosine and inosine. In contrast to GMP and IMP, AMP is not susceptible to nucleotidase activity. However, AMP can undergo conversion, through the activity of adenylate deaminase, into IMP for further degradation. Additionally, adenosine deaminase can convert adenosine to inosine for inclusion in the degradative pathway. Further catabolism of both guanosine and inosine is mediated by the common enzyme purine nucleoside phosphorylase (PNP). Guanosine is converted to guanine, whereas inosine is converted to hypoxanthine. Both guanine and hypoxanthine are subsequently converted to xanthine, by the enzymes guanine deaminase and xanthine oxidase (also known as xanthine dehydrogenase), respectively. Xanthine from either source is then converted directly to uric acid, again by the action of xanthine oxidase. As noted earlier, organisms other than humans and primates including New World monkeys possess an additional enzyme—uricase (urate oxidase), which converts uric acid to allantoic acid, a relatively soluble compound that can be further degraded to urea. Lacking this enzyme, human and primate purine metabolism ceases with the production of uric acid.18
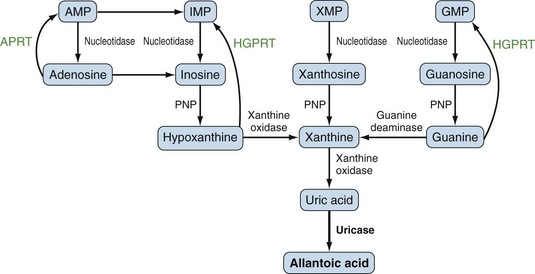
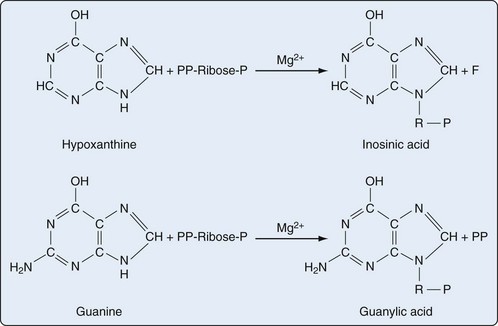
Presumably because purine synthesis is energy expensive for the cell, evolution has dictated that mechanisms exist to recover purines before they completely traverse the degradative pathway. These pathways, collectively known as purine salvage, are intimately connected to the feedback regulation of purine synthesis. The major enzyme responsible for purine salvage, hypoxanthine/guanine phosphoribosyl transferase (HGPRT), catalyzes the transfer of a phosphoribose from PRPP to either hypoxanthine or guanine, to form inosinate or guanylate, respectively (Figure 94-6). These products are then available for reinclusion in the available purine pool. A second salvage enzyme is adenine phosphoribosyl transferase (APRT), which restores adenine to adenylate. However, as described earlier, most adenosine/adenine breakdown occurs via conversion to inosinic acid. The failure of APRT deficiencies to alter uric acid production suggests that APRT plays only a minor or redundant role in purine salvage.19
Urate Overproduction: Primary and Secondary Causes
Primary Urate Overproduction
Two major variants of HGPRT deficiency have been described. Complete HGPRT deficiency, better known as Lesch-Nyhan syndrome, is an X-linked recessive disorder characterized by extremely high levels of serum urate, gouty attacks, nephrolithiasis, mental retardation, movement, and behavioral disorders including self-mutilating behavior. The disorder can occasionally arise by de novo mutation; female carriers are generally asymptomatic but may have elevated serum urate levels. In contrast to the gouty attacks and nephrolithiasis, which are direct consequences of hyperuricemia, the neurologic findings in Lesch-Nyhan syndrome are independent of hyperuricemia and unresponsive to urate-lowering drugs. Life expectancy can be greatly reduced, and these patients rarely come to the attention of adult rheumatologists.20
In contrast to Lesch-Nyhan patients, individuals with Kelley-Seegmiller syndrome have a partial deficiency of HGPRT.21 Kelley-Seegmiller patients typically present with hyperuricemia and gout and have limited or no neurologic symptoms.22 Several variants of Kelley-Seegmiller syndrome have been described, based on the extent of HGPRT inactivity and the presence/absence of neurologic findings. The mutations seen in the Kelley-Seegmiller variants tend to occur in regions of the HGPRT gene other than those identified in Lesch-Nyhan patients (whose mutations typically localize to the PRPP-binding region); whether such differences influence the presence/absence of neurologic symptoms has not been determined.23
Several hereditary defects of energy metabolism also promote hyperuricemia, mainly as a consequence of ATP consumption. Patients with glucose-6-phosphatase deficiency (type I glycogen storage, or von Gierke’s disease) demonstrate a high rate of both purine and ATP turnover. The lactic acidemia that secondarily occurs in patients with glucose-6-phosphatase deficiency may also contribute to hyperuricemia, by promoting decreased renal urate excretion (see later).24 In fructose-1-phosphate aldolase deficiency, patients lack the capacity to metabolize fructose-1-phosphate. Fructose-1-phosphate accumulation causes feedback inhibition of fructokinase and fructose accumulation in the blood. As an apparent consequence of these changes, AMP accumulates and promotes hyperuricemia by the mechanisms described earlier.25 The role of fructose intake in patients without inborn errors of fructose metabolism is discussed further later.
Secondary Urate Overproduction and Hyperuricemia
A number of secondary causes can lead to urate overproduction and hyperuricemia. In most cases, these conditions induce increased cell turnover, with resultant purine generation and breakdown. Chief among these must be counted diseases of erythropoietic, lymphopoietic, and myelopoietic cell turnover, of both malignant and nonmalignant varieties. Among the erythropoietic diseases causing hyperuricemia, autoimmune, and other hemolytic anemias (red cell destruction with increased red cell generation), sickle cell disease,26,27 polycythemia vera,28 and ineffective erythropoiesis (e.g., in pernicious and other forms of megaloblastic anemia, thalassemia, and other hemoglobinopathies) must be included.29 Patients with myeloproliferative and lymphoproliferative disorders including myelodysplastic syndrome, myeloid metaplasia, leukemias, lymphomas, and paraproteinemic diseases such as multiple myeloma and Waldenström’s macroglobulinemia are also at increased risk for hyperuricemia.30,31 In some cases, particularly in the pediatric setting, hyperuricemia and concomitant renal failure may be the first presentation of these malignancies.32 Indeed, the level of hyperuricemia may correlate with the degree of disease and cell turnover. Patients with essential thrombocytosis may also be at increased risk for hyperuricemia.33 An association between solid tumors and hyperuricemia has been reported34; given the slower turnover of solid tumor cells, solid tumor hyperuricemia tends to be less common and less severe than that seen in malignancies of bone marrow–derived cells.
Tumor lysis syndrome represents a unique form of tumor-related hyperuricemia, in which cell death induced by chemotherapy results in not only hyperuricemia but also hyperphosphatemia, hyperkalemia, and hypocalcemia, often resulting in acute renal failure and arrhythmias. Although tumor lysis syndrome occurs most commonly during treatment for hematologic malignancies, it may also occur during treatment of solid tumors.35 Although not well documented in the literature, the authors have noted hyperuricemia subsequent to the use of granulocyte colony-stimulating factor for myelofibrosis-associated anemia. Such use of colony-stimulating factors may secondarily contribute to the new onset of gout.36
Although somewhat more controversial, the increased cell turnover in patients with psoriasis has also been associated with elevated levels of serum urate.37,38 An association between sarcoidosis and hyperuricemia has also been proposed,31 again presumably relating to increased cell turnover and/or metabolic activity. However, the epidemiologic evidence supporting sarcoidosis as a cause of hyperuricemia is less than convincing.39
Conditions leading directly to the physiologic consumption/degradation of ATP also contribute to the potential for secondary purine turnover leading to hyperuricemia. Thus strenuous and prolonged exercise, particularly to levels driving anaerobic respiration, may induce transient serum urate elevations.40,41 Status epilepticus is likely to mimic these events. A number of acute illnesses including myocardial infarction and sepsis are also accompanied by ATP catabolism and may result in transient hyperuricemia.34 Patients with hereditary myopathies including metabolic myopathies such as glycogen storage disease types III, V, and VII (debranching enzyme deficiency, myophosphorylase deficiency, and muscle phosphofructokinase deficiency, respectively), as well as mitochondrial myopathies (including carnitine palmitoyltransferase deficiency and myoadenylate deaminase deficiency), are susceptible to increases in serum urate levels after even moderate exercise.34,42,43 In these individuals, a limited ability to synthesize ATP on demand apparently results in a rapid turnover of established ATP pools during exercise, with resultant purine and uric acid formation. Patients with medium-chain acyl-coenzyme A dehydrogenase deficiency, a defect of fatty acid metabolism, have also been shown to have elevated levels of serum urate, although the mechanism for this effect is not entirely clear.44
Urate Excretion: Gastrointestinal and Renal Mechanisms
Gastrointestinal Excretion of Urate
Uric acid elimination via the gastrointestinal tract has been recognized for more than 50 years but has been relatively little studied. On the basis of radiolabeled urate tracer studies, Sorensen estimated that in healthy individuals, the gastrointestinal tract is responsible for the excretion of 20% to 30% of the daily uric acid burden.45–47 Thus gastrointestinal excretion of uric acid may represent a minor pathway for urate excretion under most circumstances. Gastrointestinal uric acid excretion may become more important in settings of renal insufficiency, however, particularly in view of animal studies suggesting that uric acid excretion via the gut may increase in a compensatory manner in the setting of renal failure and decreased renal uric acid excretion.48 Mechanisms of uric acid transport into the gut appear to include exocrine secretion (saliva, gastric, and pancreatic juices), as well as direct bowel secretory mechanisms.34 Uric acid is apparently excreted into the gut in its native form and then undergoes degradation by intestinal flora.47
Renal Excretion of Uric Acid: Normal Mechanisms
Subsequent to ultrafiltration, urate (initially in a monovalent ion form) undergoes several distinct handling steps: (1) a resorption step, in which as much as 90% to 98% of the filtered urate undergoes reclamation; (2) a secretion step, in which most of the urate resorbed in step 1 is retransferred back into the tubule lumen; and (3) a possible additional resorption step in which a smaller amount of uric acid is then resorbed. The net result is excretion of approximately 10% of the filtered load. In fully functioning nephrons, these steps are responsive to serum urate levels, such that rises in serum urate induce increased renal excretion and maintenance of total body urate homeostasis. Early studies, particularly in mouse models, suggested that these three steps might occur in anatomic sequence, in the proximal tubule, descending loop of Henle, and distal convoluted tubule, respectively. However, studies in humans over the past decade including both genetic and physiologic approaches suggest that these functions are likely to overlap and to occur mainly in the proximal tubule. Moreover, these same studies have emphasized the importance of organic anion transporters (OATs) and other active and passive transport molecules in the movements of urate in both directions across the proximal tubule (Figure 94-7).49,50
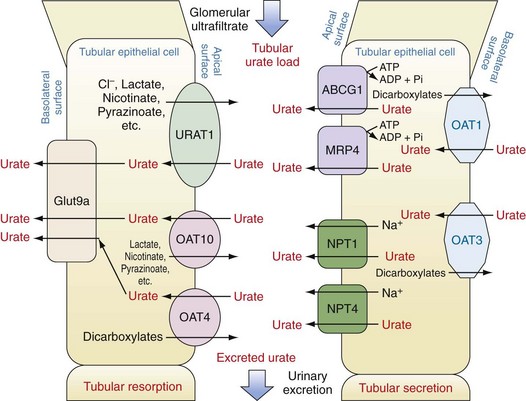
Urate Resorption.
Urate resorption in the proximal tubule depends on the action of several apical surface transporters and at least one resorption transporter at the basolateral surface (see Figure 94-7). In humans, the most important of the apical-surface transporters appears to be URAT1 (gene, SLC22A12). URAT1 acts as a urate/anion exchanger to transfer urate from the tubule lumen to the epithelial cytosol. The major inorganic counter-ion for URAT1 activity is Cl−. However, organic anions such as lactate, pyrazinoate, and nicotinate can substitute for chloride, with potential clinical consequences as discussed later. The importance of URAT1 to renal urate resorption is indicated by the fact that patients with inactivating mutations of URAT1 excrete nearly 100% of their filtered urate and demonstrate low serum urate levels (along with increased urinary uric acid levels and risk for uric acid kidney stones).51–53 Moreover, several well-established urate-lowering drugs including probenecid, benzbromarone, and losartan act by inhibiting URAT1. Conversely, other mutations in the URAT1 gene appear to convey a risk for increased urate resorption and hyperuricemia, presumably through a gain-of-function mutation.
OAT4 (gene, SLC22A11) and OAT10 (gene, SLC22A8) are two other apical anion transporters involved in renal uric acid resorption. Like URAT1, OAT10 is an anion exchange transporter; counter-ions that can promote urate transport by OAT10 include lactate, pyrazinoate, and nicotinate, a fact of clinical importance (see later). In contrast, although OAT4 also transports urate from the tubular lumen to the cytosol of renal epithelial cells, the counterions it employs tend to be dicarboxylates.49,54
Urate transport by URAT1, OAT4, and OAT10 would lead to accumulation of urate intracellularly and presumably to gradients that would eventually impair further urate uptake if a mechanism did not exist to transport intracellular urate out of the cell at the basolateral surface. This function appears to be served by Glut9a (gene, SLC2A9; also confusingly known as URATv1), which was first identified as a glucose transport family protein but has little or no glucose transport capacity. Instead, Glut9a is an effective transporter of urate from the renal epithelial cell out into the renal interstitium.55,56 A number of different inactivating mutations of Glut9a have been identified in both humans and mice; in each case the result is impaired urate resorption, increased urate excretion, and hypouricemia.55,57–61 Glut9a and its splice variant, Glut9b, are also expressed on other cells including chondrocytes, leukocytes, intestinal cells, and hepatocytes; the role of Glut9 proteins on these cells is actively being explored.62,63
Urate Secretion.
Other transport proteins in the renal tubule epithelium regulate the excretion of urate from the peritubular fluid into the tubular lumen (see Figure 94-7). At the basolateral surface, OAT1 and OAT3 (genes, SLC22A6 and SLC22A8, respectively) transport urate from the interstitium into the epithelial cell cytosol. These transporters act via exchange with dicarboxylate anions and transport not only urate, but other organic anions and some drugs.64 At the apical surface of proximal tubule cells, two proteins have been identified that serve as urate-extruding transporters. The multidrug resistance protein MRP4 (gene, ABCC4) mediates ATP-dependent urate transport.65 Whether energy failure promotes hyperuricemia by impairing MRP4 has not been established but would seem plausible. ABCG2 (gene, ABCG2) also directly mediates urate excretion.66,67 Genetic association studies have implicated two other anion transport proteins as playing a role in apical urate transport outside of the cell, namely, NPT1 (gene, SCL17A1) and NPT4 (gene, SLC17A3).66,68–70 Additionally, genetic studies have implicated nontransporter proteins as playing roles in urate excretion including PDZK1, CARMIL, NHERF1, SMCT1, SLC5A8, SMCT2, and SLC2A12. It is thought that some of these proteins may contribute to a macromolecular complex regulating urate transport.71
Renal Causes of Hyperuricemia
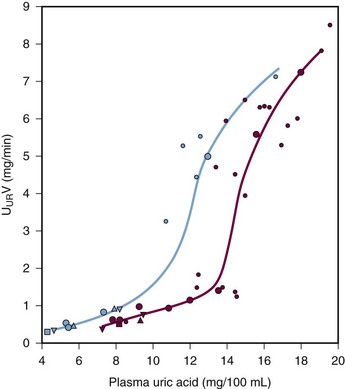
Many patients with hyperuricemia underexcrete urate; that is, for any given serum urate level, their degree of renal urate excretion is inadequate and less than is seen in normal controls (Figure 94-8). The mechanisms of urate underexcretion are various and stem from either primary or secondary renal effects.
Primary Urate Underexcretion
In a subset of patients, hereditary defects of renal tubule urate excretion result in hyperuricemia. With the identification of the aforementioned renal urate transporters, the underlying basis for some of these defects has become apparent. For example, up to 10% of gout cases in white Europeans may be attributable to hyperuricemia induced by defects in the urate exporter ABCG2.66,67 Gain-of-function mutations of other transport-related proteins (e.g., PDZK1, CARMIL, NHERF1) may actually promote tubular urate resorption mediated by URAT1 activity. Patients with such intrinsic tubular defects leading to net urate underexcretion frequently display normal renal filtration and normal serum creatinine levels.
Familial juvenile hyperuricemic nephropathy (FJHN), also known as medullary cystic kidney disease (MDCK), constitutes a group of autosomal dominant disorders characterized by early hyperuricemia and progressive chronic kidney disease.72 The hyperuricemia typically precedes the renal insufficiency and is considered to be primary. Three variants are recognized, designated MDCK 1, 2, and 3. In MDCK2, mutations in the uromodulin gene result in underproduction and/or misproduction of uromodulin (Tamm-Horsfall protein), the most prevalent protein secreted by the kidney.73–75 The probable importance of uromodulin deficiencies to FJHN has recently been underlined by the observation that although patients with the MDCK1 and 3 subtypes of FJHN have mutations that are not in the uromodulin gene, they nevertheless display a phenotype of decreased uromodulin expression.76 The mechanisms through which uromodulin deficiencies predispose to hyperuricemia are not yet understood, but mice transgenic for human uromodulin mutations develop renal tubular abnormalities and concentrating defects.77
Secondary Causes of Renal Urate Underexcretion
A large number of underlying causes can result in nephrogenic retention of urate and subsequent hyperuricemia. These include acute or chronic renal failure, the effects of toxins and drugs, and systemic illnesses that alter renal urate handling directly or indirectly.78
Age and Gender.
Classic studies by French and colleagues79 documented that serum urate levels tend to be low in children. In males, urate levels increase precipitously at puberty, a time when females experience only a slight increase in serum urate. For women, a gradual increase over the subsequent years, followed by another rise at menopause, finally brings the serum urate near to that of men, consistent with Hippocrates’ astute aphorisms that “a young man does not take the gout until [around the time] he indulges in coition” and that “a woman does not take the gout, unless her menses be stopped.”79,80 This discrepancy between men and women, in the period between puberty and menses, suggests strongly that sex hormones play a role in urate regulation. Indeed, studies suggest that in women, estrogenic hormones may promote renal urate excretion.81 Conversely, an active role for androgens in promoting hyperuricemia may be inferred from Hippocrates’ assertion that “Eunichs do not take the gout” and from studies indicating that androgens and estrogens have opposing effects on renal organic anion transporters.82 In pregnant women, increases in serum urate levels are characteristic of preeclampsia, a reproductive emergency consisting of hypertension and proteinuria. The hyperuricemia of pre-eclampsia is thought to result from that condition’s renal dysfunction; hyperuricemia in pre-eclampsia does not lead to gout but is considered by some investigators to secondarily contribute to pre-eclamptic renal dysfunction.83
Systemic Illnesses (Table 94-1).
Table 94-1 Systemic Conditions Promoting Hyperuricemia
| Overproduction |
| Underexcretion |
| Both Overproduction and Underexcretion |
| Metabolic States |
Dehydration (volume depletion) on any basis promotes hyperuricemia.84 Once again, the mechanisms are complex but include decreased renal perfusion and subsequently decreased urate filtration and delivery to the proximal tubule. Subsequent sodium retention will reduce tubular urate secretion, probably at the Na+/urate co-transporters NPT1 and NPT4. Patients exposed to low-sodium diets may also retain urate in an effort to retain sodium.
Several metabolic and/or endocrinologic conditions have been associated with hyperuricemia, although whether these represent independent associations has not been determined. These include hypothyroidism and hyperthyroidism, and hypoparathyroidism and hyperparathyroidism. Obesity is associated with hyperuricemia,85 and weight loss has been shown to reduce both serum urate levels86 and risk of gout.87 Although it is conceivable that adiposity itself may promote hyperuricemia, this relationship is complex because adiposity may also reflect diet and thyroid status. Patients who have undergone renal transplant experience increases in prevalence of hyperuricemia and gout (2% to 13%). It is likely that the hyperuricemia in renal transplant patients is not related to the transplant per se, but to other factors such as intrinsic renal insufficiency, diuretic use, and especially the use of cyclosporine to suppress rejection (discussed further later).88 Reciprocally, some studies suggest that hyperuricemia in transplant patients may contribute to worsening renal function.
Drugs (Table 94-2).
Diuretics are among the most commonly used agents to treat hypertension and congestive heart failure, and it has long been appreciated that many diuretics promote hyperuricemia and subsequent gout.89,90 Despite early assessments suggesting otherwise,91 the increase in gout risk owing to diuretic use is substantial and may range as high as 3- to 20-fold.92 The mechanisms through which diuretics raise serum urate are incompletely elucidated but include the induction of sodium wasting and volume depletion, with a resultant decrease in the fractional excretion of urate.93 However, individual diuretics may also have more specific and direct effects on renal urate handling. For example, loop diuretics such as furosemide and bumetanide interact directly with the tubular urate transporter NPT4,70 and both thiazide and loop diuretics inhibit the renal urate exporter MRP4.94 Indeed, despite the volume-depleting effects of diuresis, not all diuretics promote hyperuricemia. For example, the potassium-sparing diuretics triamterene, amiloride, and spironolactone do not raise urate. Interestingly, some diuretics actually lower serum urate levels, apparently by directly promoting renal urate excretion even as they induce volume depletion. One such drug was tienilic acid, an effective diuretic and antihyperuricemic that was withdrawn from use owing to hepatotoxicity.
Table 94-2 Drugs Promoting Hyperuricemia
| Diuretics |
| Organic Acids |
| Other |
Several drugs that are weak organic acids may raise serum urate by serving as counter-ions to promote urate retention by URAT1 and OAT10. These drugs have also been assumed to inhibit tubular secretion, possibly by acting as urate competitors. Among these agents is the lipid-lowering drug nicotinic acid, which may not only block urate secretion but also promote urate formation.90 Low-dose salicylates including the low doses of aspirin used for cardioprotection may also raise urate by impairing renal urate efflux.94 At high doses salicylates become uricosuric, apparently through the inhibition of URAT1.95 The antituberculous agent pyrazinamide is the most potent urate-retaining agent known.90 Pyrazinamide is metabolized to pyrazinoate and subsequently to 5-hydroxypyrazinoate96; it is likely that these organic anions act in a manner similar to nicotinate and salicylate. Another antituberculous agent, ethambutol, can also reduce renal tubular urate excretion and promote hyperuricemia. However, the mechanism of ethambutol’s action is not well established.97
The immunosuppressant cyclosporine is well known to promote decreased renal urate excretion and hyperuricemia. The mechanism of cyclosporine-induced hyperuricemia is presumed to depend, at least in part, on the common cyclosporine effect of decreasing renal glomerular filtration; reciprocally, hyperuricemia may exacerbate the nephrotoxic cyclosporine effects.98 However, tacrolimus, whose mechanism of immune action is not dissimilar to that of cyclosporine, and which also can impair renal function, does not promote similar hyperuricemia. Moreover, cyclosporine’s effect on urate retention appears to exceed its effects on renal filtration, suggesting a probable direct effect on tubular urate transport.90,99 Most studies of cyclosporine’s effects on urate have been performed in patients who have undergone renal transplant; cyclosporine’s effects on urate in other settings are less well established.
Toxins.
Several toxins can affect the kidney to promote hyperuricemia. Chief among these is lead. Lead exposure is endemic to Western society, but there have been a number of periods during which lead exposure may have been excessive (e.g., during the Roman Empire). A connection between lead exposure, hyperuricemia, and gout has been suspected at least since the 18th century.100 In the 20th century, a large cohort of patients with lead-induced hyperuricemia (saturnine gout) was recognized during and after the era of Prohibition, predominantly in the Southeastern United States and relating to the home brewing of whiskey (moonshine, or white lightning) using lead-lined vessels (typically, automobile radiators). Lead consumption results in distribution to a reservoir in bone and may have adverse effects on the central nervous system as well. In the kidney, lead poisoning leads to interstitial and perivascular fibrosis, as well as glomerular and tubular degeneration.101,102 Although patients with lead nephropathy do experience mild to moderate renal insufficiency, their clearance of urate is excessively limited, indicating a tubular defect in urate excretion.103 Suggestions that lead exposure may also promote purine turnover are provocative but have been less well supported.104 Although saturnine gout is currently a relatively rare condition, epidemiologic data suggest that it may be more prevalent than commonly assumed.105 In patients with moonshine nephropathy, associated lifestyle factors (e.g., alcohol consumption, obesity) may play a significant role in the genesis of the hyperuricemia.106
Patients with chronic beryllium poisoning, usually as a result of an occupational exposure, may also suffer diminished renal urate excretion and hyperuricemia.107

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


