23 Complement System
Historical Aspects
Today, complement is a collective term designating a group of plasma and membrane proteins that play a key role in innate and adaptive immunity. The complement system is ancient, predating chordates such as the lamprey and hagfish.1,2 Complement was discovered in the late nineteenth century by experimental pathologists attempting to understand the protective basis of vaccination and the cause of transfusion reactions.3 In these studies, it became apparent that the cell-free portion of blood contained a lytic substance for bacteria and for transfused red blood cells (RBCs). This “factor” was heat labile (destroyed by heating at 56° C for 30 minutes), unstable (it lost activity if serum was left on the bench top overnight), and innate or nonspecific (all individuals had this lytic substance). It was contrasted to antibody, which was heat stable, and only animals immunized to the specific bacteria contained this factor. Through mixing experiments, these two factors were found to be required for lysis—a specific recognition piece (antibody) and a nonspecific lytic piece (complement). In retrospect, this was the discovery of the classical complement pathway and led to its characterization as a heat-labile, bactericidal factor that “complemented” the acquired factor raised by immunization. Similarly, in the blood of patients who recovered from transfusion reactions, a heat-stable substance (antibody) was found that recognized the transfused erythrocytes. However, it did not lyse the transfused cells; rather, it cooperated with a second lytic substance (complement) present in fresh serum.
These historical points account for why lysis became linked to the complement system and why the study of antibody and complement dominated the field of immunology for the next half century. Of interest, the first autoimmune disease to be recognized was only a few years later. It was a hemolytic anemia (paroxysmal cold hemoglobinuria) in which the same pair of reactants, antibody and complement, were shown to mediate RBC lysis.4 Consequently, this double-edged sword aspect of the complement system was recognized early, but not until the 1960s was there such a clear-cut demonstration of its protective versus injurious role. Compared with normal mice, C5-deficient mice were 100-fold more sensitive to death from pneumococcal infection but 100-fold less sensitive to tissue destruction by autoantibodies. Even more to the point, we now know that inherited deficiencies of C1, C4, and C2 strongly predispose to SLE, yet immune complex (IC) deposition and complement activation contribute to tissue damage in SLE.
Function
A major activity of the complement system is to modify membranes and tag antigens through covalent attachment of its activation fragments (Figure 23-1). For example, several million C3b molecules can bind to a bacterial surface in less than 2 minutes. One goal of these deposited complement proteins is to opsonize the target. C3b (and its degradation fragments) and C4b are ligands for complement receptors on peripheral blood cells, tissue macrophages, and antigen-presenting cells such as follicular dendritic cells. Complement ligands and receptors are particularly adept at this coupling process, known as immune adherence. On phagocytic cells, this often leads to ingestion of the complement-coated infectious particle, IC, or cellular debris. Additionally, microorganisms (e.g., gram-negative bacteria) and host cells may be lysed by the terminal complement components (C5b-C9). Many microbes, though, possess capsules that make them resistant to lysis (e.g., most gram-positive organisms). In human diseases featuring autoantibodies, cells and tissues are similarly opsonized and membrane integrity can be compromised. A recent extension of this opsonic function relates to complement’s role in the clearance of cellular debris following necrosis or apoptosis. In particular, one hypothesis to explain autoantibody formation in SLE is the complement system’s failure to properly clear or handle apoptotic cells.5
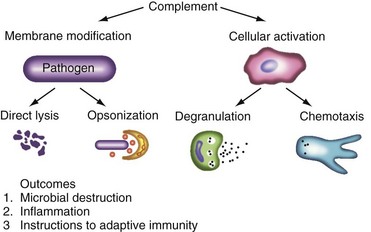
A second function of the complement system is cellular activation.6 It prepares the local environment for defense against infection (see Figure 23-1). In the proteolytic steps of the early complement activation process, mediators that activate nearby cells are released. These low-molecular-weight fragments (C4a, C3a, C5a) are termed anaphylatoxins because, if released in excessive amounts, they induce shock. C3a and C5a, the two most powerful mediators, bind to their respective receptors at sites of complement activation. This causes histamine release by mast cells and chemotaxis to the area by phagocytic cells. With improved reagents and newer technology, receptors for C3a and C5a have now been shown to be much more widely expressed than initially thought; for example, they are expressed by epithelial cells, T cells, hepatocytes, endothelial cells, neurons, and other cell types. C3a and C5a may engage their receptors on endothelial and neuronal cells to alter blood flow and modulate cellular function.
Nomenclature
There are three activation cascades: the classical pathway (CP), the alternative pathway (AP), and the lectin pathway (LP) (Figure 23-2). They converge to activate C3 and then C5, leading to opsonization and membrane perturbation.
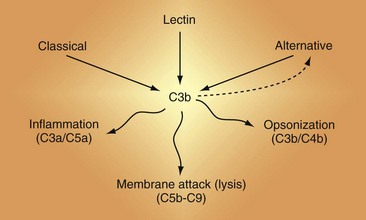
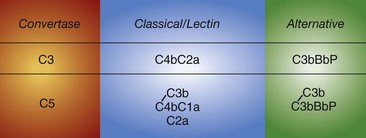
The nine proteins of the CP are designated by the uppercase letter C, followed by a number (Table 23-1). These components act in numeric order, except for C4, which is cleaved in parallel with C2 and before C3. Components of the AP are designated by capital letters (e.g., factor B). Regulatory proteins are designated by a descriptive title (e.g., C4 binding protein) or a letter (e.g., factor H) (Table 23-2). Single components or multimeric complexes that have enzymatic activity are designated by a bar (e.g., 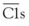 ). The loss of hemolytic potential by a component is usually designated by a lowercase prefix (e.g., iC3b). Fragments generated during complement activation are designated by a lowercase letter suffix (e.g., C3a, C3b). Except for the fragments of C2 (which are the opposite), the “a” fragment is liberated into the surrounding milieu, whereas the “b” fragment becomes cell bound and continues the cascade.
). The loss of hemolytic potential by a component is usually designated by a lowercase prefix (e.g., iC3b). Fragments generated during complement activation are designated by a lowercase letter suffix (e.g., C3a, C3b). Except for the fragments of C2 (which are the opposite), the “a” fragment is liberated into the surrounding milieu, whereas the “b” fragment becomes cell bound and continues the cascade.
Table 23-1 Components of the Complement Activation Cascades
| Component | Serum Concentration (µg/mL) | Function |
|---|---|---|
| Classical Pathway (CP) | ||
| C1 | 50 | Binds IC to trigger CP |
| C1q subcomponent | Binds Fc portion of IgG, IgM | |
| C1r subcomponent | Protease: cleaves C1s | |
| C1s subcomponent | Protease: cleaves C4 and C2 | |
| C4 | 200-500 | Opsonin* (C4b), C4a† |
| C2 | 20 | Protease: cleaves C3 and C5 as part of the convertases |
| Lectin Pathway (LP) | ||
| MBL | 150 (wide range) | Binds sugars to trigger LP |
| MASP-1/3 | 5 | Proteases |
| MASP-2 | 5 | Protease: cleaves C4 and C2 |
| Alternative Pathway (AP) | ||
| Factor D | 1-2 | Protease: cleaves factor B |
| Factor B | 150-250 | Protease: cleaves C3 and C5 as part of the convertases |
| Properdin | 25 | Stabilizes AP convertases |
| Central protein | ||
| C3 | 550-1200 | Opsonin† (C3b), C3a†; convertases |
| Terminal Pathway (TP) | ||
| C5 | 70 | MAC component (C5b), C5a† |
| C6 | 60 | MAC component |
| C7 | 60 | MAC component |
| C8 | 60 | MAC component (pore formation) |
| C9 | 60 | MAC component (pore formation) |
IC, immune complex; Ig, immunoglobulin; MAC, membrane attack complex; MASP, mannose-binding associated serine protease; MBL, mannose-binding lectin.
* C4b forms part of the CP C3 and C5 convertases. It anchors the enzyme complex to the target. Likewise, for the AP C3 and C5 convertases, C3b anchors these enzyme complexes to the target.
† C4a, C3a, and C5a are liberated on cleavage of C4, C3, and C5.
Activation Cascades
Because many rheumatic diseases feature autoantibodies and therefore ICs, an understanding of the general principles of the activation cascades is essential to characterizing complement’s role in a disease such as SLE3,7–10 (Figure 23-3; see also Figure 23-2).
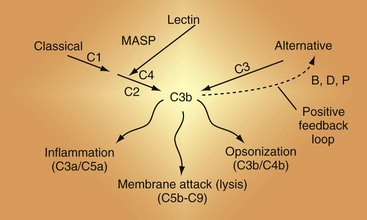
Figure 23-3 The complement activation pathways. Note that C4 and C2 serve identical purposes in the classical pathway (CP) and the lectin pathway (LP). They are cleaved by mannan-associated serine protease-2 (MASP-2) in the LP and by C1 in the CP. The convertases that cleave C3 are shown in Figure 23-6. C1 connects immune complexes to the CP as the C1q subcomponent binds to the Fc of antibody and the C1s subcomponent cleaves C4 and C2. In the LP, the lectin carries a MASP that cleaves C4 and C2.
Classical Pathway
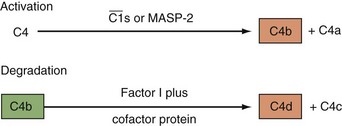
The CP is activated primarily by ICs and a few other substances including C-reactive protein.11 Human IgM and IgG subclasses 1 and 3 efficiently activate the CP. Once C1 is bound through the Fc portion of IgG or IgM interacting with the C1q subunit of the C1 complex (Figure 23-4), the C1r enzymatic subcomponent autoactivates by proteolysis and then cleaves Cls to 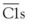 . C1s cleaves C4 to C4b, releasing the C4a fragment (Figure 23-5). The C4b fragment, having had its thioester bond disrupted, now has the transient ability (a few microseconds) to covalently bind to a hydroxyl or amino group on a nearby target. C2 is also a substrate for C1, and C2 is cleaved by C1 to yield C2a and C2b. C2a interacts with 10% to 20% of the target-bound C4b to form the CP C3 convertase C4bC2a (Figure 23-6). The enzymatic or catalytic domain of this complex is C2a, which cleaves C3 to form C3a and activated C3b (Figure 23-7). Activated C3b, like C4b, has a few microseconds to attach to a target by binding covalently to a hydroxyl group (analogous to the interaction of C4b with a hydroxyl or amino group), where it serves as the major opsonin of the complement system. In addition, some of the newly activated C3b binds covalently to a specific region on the C4b component of the C3 convertase to form the CP C5 convertase, C4bC2aC3b (see Figure 23-6).
. C1s cleaves C4 to C4b, releasing the C4a fragment (Figure 23-5). The C4b fragment, having had its thioester bond disrupted, now has the transient ability (a few microseconds) to covalently bind to a hydroxyl or amino group on a nearby target. C2 is also a substrate for C1, and C2 is cleaved by C1 to yield C2a and C2b. C2a interacts with 10% to 20% of the target-bound C4b to form the CP C3 convertase C4bC2a (Figure 23-6). The enzymatic or catalytic domain of this complex is C2a, which cleaves C3 to form C3a and activated C3b (Figure 23-7). Activated C3b, like C4b, has a few microseconds to attach to a target by binding covalently to a hydroxyl group (analogous to the interaction of C4b with a hydroxyl or amino group), where it serves as the major opsonin of the complement system. In addition, some of the newly activated C3b binds covalently to a specific region on the C4b component of the C3 convertase to form the CP C5 convertase, C4bC2aC3b (see Figure 23-6).
CP activation by polyclonal antibodies is efficient on membranes of cells, bacteria, and viruses. In one illustrative study, complement activation by IgG binding to a membrane antigen of a nucleated cell produced about 2.5 million bound C4b molecules, and about 0.5 million C2a molecules subsequently attached to these C4bs to form convertases.12 In less than 5 minutes, 21 million (representing a 10× amplification over the number of attached C4b molecules) C3b molecules were deposited. Further, only about 10% to 20% of the C4b and C3b molecules generated bound to the target. The rest were inactivated by hydrolysis in the fluid phase and then degraded by fluid-phase inhibitors. Concomitantly, C4a, C3a, and C5a anaphylatoxins were generated equal to the number of C4b, C3b, and C5b fragments produced. Approximately 1 million membrane attack complexes (MACs) were formed. Interaction of the complement system with soluble antigens is a less efficient process, particularly the formation of the C3 and C5 convertases.
Lectin Pathway
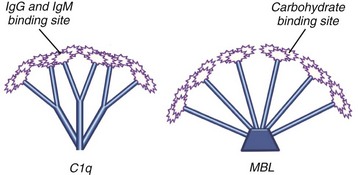
Lectins are carbohydrate-binding proteins5,13–15 (see Figure 23-4). They were initially described as proteins capable of agglutinating RBCs. They are important players in innate immunity and in rheumatic diseases. In particular, mannose-binding lectin (MBL) is a hepatocyte-synthesized plasma protein that preferentially binds to repeating mannoses and certain other repeating oligosaccharides of pathogens. MBL resembles C1q (it belongs to the same family of proteins, known as collectins), consisting of an oligomer with a terminal collagenous domain on one end and a globular domain on the other (see Figure 23-4). The main difference is that the carboxy terminus of MBL possesses a carbohydrate-recognition domain, whereas C1q has an Ig-binding domain. Like C1q binding to the Fc portion of IgG, MBL engagement with its sugar ligand leads to activation of serine proteases similar to C1r and C1s, called mannose-associated serine proteases (MASPs). MASP-2 cleaves C4 and C2. MBL was discovered during an investigation into the cause of bacteremia in young children. MBL deficiency may be a predisposing factor for SLE and RA and contribute to a more severe disease phenotype.
The LP shares many features with the CP.8,9,13,14 The differences occur during the first few steps when MBL binds to an activating surface such as bacteria with the appropriate repeating oligosaccharides. Each element of the MBL-MASP complex is structurally and functionally homologous to the corresponding elements in the CP. C1q and MBL have globular ends through which they attach to their respective substrates. The MASP-2 in the complex cleaves C4 to form C4a and C4b (see Figure 23-5) and C2 to form C2a and C2b—identical to C1 cleaving C4 and C2. The C4b2a complex is the same as the C3 convertase generated in the CP (see Figure 23-6). The LP is a key player in innate immunity, where MBL and related lectins such as surfactants and ficolins bind to their cognate residues on microbes to trigger complement activation. At one time, MASPs (and possibly C1) directly activated C3, but through the evolution of C4 and C2, a more efficient C3 activating process was engineered.14
Alternative Pathway
The AP was the second complement activation pathway discovered (hence the name), but it is phylogenetically the most ancient.1,2 In contrast to the CP and LP, the AP does not require antibody or a lectin to be triggered. It likely plays a larger role in rheumatic diseases than previously appreciated because of its so-called feedback or amplification loop.16 Thus if C3b is deposited on a target by the CP or LP, the AP can enhance C3b deposition many-fold. The AP is also continuously active at a low level (like an idling car). The plasma inhibitors factor H and factor I and host cell surface regulators keep the system in check. In the absence of these regulators, as exemplified by inherited deficiencies, the system fires to exhaustion. The AP routinely “kicks into gear” in the setting of microbes. If C3b deposits on an activating surface (i.e., one that lacks regulators), the system’s feedback loop allows several million C3b molecules to be deposited in a few minutes. In this pathway, factor B, structurally and functionally homologous to C2, binds to the deposited C3b, and is then cleaved by the active factor D to Bb and Ba. C3bBb is the AP C3 convertase (see Figure 23-6). It is stabilized by properdin (P), increasing the half-life of the enzyme 5- to 10-fold. The catalytic serine protease Bb of the C3bBbP enzyme complex then cleaves C3 to C3a and C3b; thus a feedback loop is set up. Some of the newly generated C3b molecules bind covalently to C3b already deposited on the target to form a C5 convertase (C3b2Bb). In vivo, the C3 and C5 AP convertases are short-lived owing to spontaneous decay, unless stabilized by properdin. Two new developments in the AP are that (1) MASP 1/3 appears to be required to activate pro-factor D to factor D17 and (2) properdin may bind to certain targets to then serve as a platform to initiate the AP.18
Regulators
Physiologic Regulation
Unregulated, the complement system would fire to exhaustion, as illustrated by a complete deficiency of plasma regulatory proteins (factor H or factor I).19–21 The complement system has evolved to allow unimpeded activation on a microbe but to limit the process in time and space. The reaction must be finite in time to avoid excessive consumption of components in any one reaction and finite in space to minimize damage to surrounding host tissue. Thus a typical complement reaction on a microbial target occurs within a few minutes, and during this time, self-tissue is protected by plasma and membrane regulators. Many of these inhibitors act at the critical step of convertase formation. The convertase enzyme complexes intrinsically have short half-lives, and many of the inhibitors act to prevent their formation or to dissociate (decay) already formed complexes.
Fluid-Phase and Membrane Inhibitors
These regulators function at the initiating step (see Figure 23-3), the formation of the C3 and C5 convertases (see Figure 23-6), and the insertion of the MAC. The plasma inhibitors prevent fluid-phase activation (no target), whereas the membrane inhibitors prevent activation on healthy self-tissue (wrong target). The plasma inhibitors may also bind to damaged cells and extracellular matrices, where they now serve as a membrane regulator.8,9,22,23
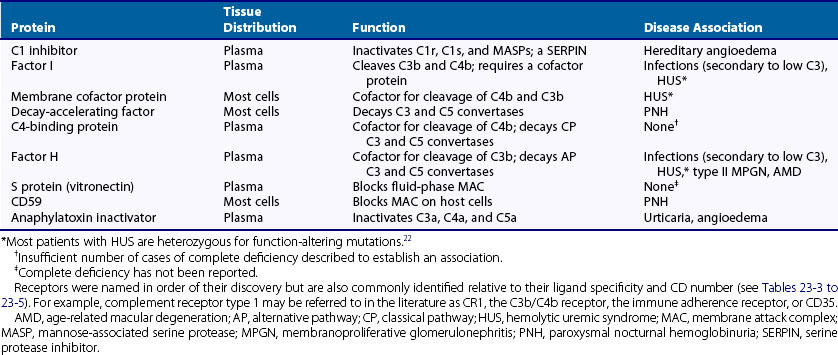
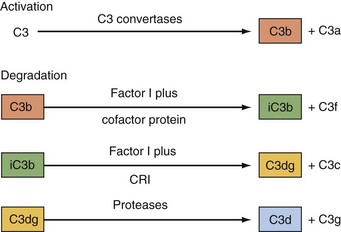
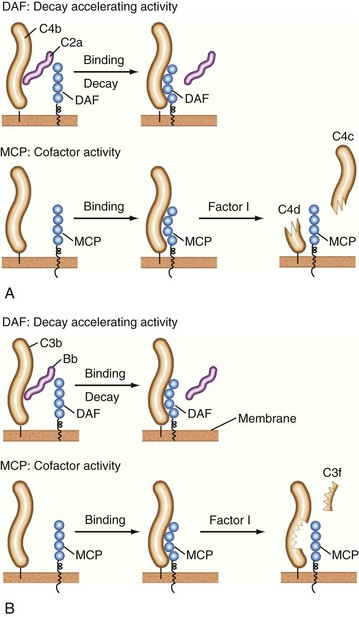
C1 inhibitor, a serine protease inhibitor (SERPIN), binds to C1r and C1s and to MASPs. The C1 complex is thereby disrupted, leaving C1q bound to antibody in the IC. C1q may then interact with C1q receptors to facilitate clearance of the IC.24,25 C1 inhibitor also prevents excessive and chronic fluid-phase C1 activation.26 The C3 and C5 convertases are regulated by a family of proteins that include the membrane proteins decay-accelerating factor (DAF or CD55) and membrane cofactor protein (MCP or CD46) and the serum inhibitors C4-binding protein and factor H.8–10,19–21 These proteins act in three ways: prevent convertase assembly, disassemble convertases (decay-accelerating activity; Figure 23-8A), and serve as cofactors for the proteolytic inactivation of C4b and C3b (cofactor activity; Figure 23-8B). The cleavage is mediated by the plasma serine protease factor I. The MAC is also regulated by plasma and cell-anchored protein. CD59 is a widely expressed glycolipid-anchored membrane protein that binds C8 and C9 to prevent their membrane insertion, while the plasma protein vitronectin (S protein) inactivates fluid-phase MAC.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


