80 Clinical Features of Systemic Lupus Erythematosus
Lupus nephritis is the most common of the potentially life-threatening manifestations.
Atherosclerosis, a complication of long-standing SLE, requires aggressive risk factor modification.
Classification Criteria
Systemic lupus erythematosus (SLE) is the prototypic systemic autoimmune disease characterized by diverse multisystem involvement and the production of an array of autoantibodies. Clinical features in individual patients can be quite variable, ranging from mild joint and skin involvement to severe life-threatening internal organ disease. Criteria for the classification of SLE were initially developed by the American College of Rheumatology (ACR) in 1971, revised in 1982, and revised for a second time in 19971,2 (Table 80-1). A person must fulfill 4 of 11 criteria to be classified as SLE, all other reasonable diagnoses having been excluded. A patient does not have to manifest all 4 criteria simultaneously; the required 4 of 11 criteria can be fulfilled over a period of weeks or years. The ACR criteria were developed as a means of classifying patients with SLE for the purpose of inclusion in clinical and epidemiologic studies. In clinical practice, these criteria are often cited to support a diagnosis of SLE. However, it should be emphasized that fulfillment of these classification criteria is not an absolute requirement for diagnosis. Rather, diagnosis typically rests on the judgment of an experienced clinician who recognizes a characteristic constellation of symptoms and signs in the setting of supportive serologic studies, after exclusion of alternative differential diagnostic possibilities. Recently, a concerted effort has been made to further revise the classification criteria, for example, to make lupus nephritis a “stand-alone” criterion, to increase the weight of neurologic manifestations, and/or to add a low-complement criterion. These proposed changes to the classification criteria are currently in development.
Table 80-1 1997 Update of the 1982 Revised American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus*
| Criterion | Definition |
|---|---|
| Malar rash | Fixed erythema, flat or raised, over the malar eminences, sparing the nasolabial folds |
| Discoid rash | Erythematous raised patches with adherent keratotic scale and follicular plugging; atrophic scarring may occur in older lesions |
| Photosensitivity | Skin rash as a result of unusual reaction to sunlight, by patient history or physician observation |
| Oral ulcers | Oral or nasopharyngeal ulceration, usually painless, observed by a physician |
| Arthritis | Nonerosive arthritis involving two or more peripheral joints, characterized by tenderness, swelling, or effusion |
| Serositis | |
| Renal disorder | |
| Neurologic disorder | |
| Hematologic disorder | |
| Immunologic disorder | a. Anti-DNA: antibody to native DNA in abnormal titer or b. Anti-Smith: presence of antibody to Sm nuclear antigen or c. Positive finding of antiphospholipid antibodies based on (1) abnormal serum concentration of IgG or IgM anticardiolipin antibodies, (2) positive test result for lupus anticoagulant using a standard method, or (3) false-positive serologic test for syphilis known to be positive for at least 6 mo and confirmed by Treponema pallidum immobilization or fluorescent treponemal antibody absorption test |
| Positive antinuclear antibody | An abnormal titer of antinuclear antibody by immunofluorescence or an equivalent assay at any point in time and in the absence of drugs known to be associated with drug-induced lupus syndromes |
* The presence of four or more criteria is required for systemic lupus erythematosus classification. Exclude all other reasonable diagnoses.
Epidemiology
Prevalence and incidence rates of SLE vary widely in the literature. Reported prevalence frequencies range from 20 to 240 per 100,000 persons, and reported incidence rates range from 1 to 10 per 100,000 person-years.3 This variation is partly due to methodological differences between studies (e.g., different case definitions of SLE and methods of case ascertainment). One study from Rochester, Minnesota, determined that the incidence of SLE increased almost fourfold between the time periods of 1950 to 1979 and 1980 to 1992.4 This increase in reported incidence may reflect a combination of factors, including an actual increase in disease, changes in population demographics, more widespread case-finding efforts, and detection of milder cases.
Prevalence and incidence of SLE vary across gender, geographic regions, and racial/ethnic groups. SLE demonstrates a striking female predominance with a peak incidence during reproductive years. The degree of female predominance varies with age. The female-to-male ratio is 10 to 15 : 1 in adults, 3 : 1 in older-onset SLE, and 8 : 1 in children.5 The prevalence of SLE is believed to be approximately three- to fourfold higher in African-American, Asian, and Hispanic populations compared with white populations.6 SLE is rare among blacks in Africa.
Most SLE patients present with their disease between 15 and 64 years of age.7 Patients with pediatric-onset SLE (<16 years old) are more likely to be African-American than those with later-onset SLE.7 SLE tends to be more severe in men and in pediatric patients. Late-onset SLE (>50 years old) is characterized by a more insidious onset with a higher occurrence of serositis and pulmonary involvement and a lower incidence of malar rash, photosensitivity, alopecia, Raynaud’s phenomenon, neuropsychiatric disease, and nephritis.7
Clinical Features
The great variability in the expression and severity of SLE constitutes a major challenge to accurate diagnosis. Although it is difficult to pinpoint their precise frequencies, it is clear that the most common presenting manifestations are constitutional symptoms (fever, fatigue, and/or weight loss), cutaneous manifestations (e.g., malar rash), and articular manifestations (arthritis and/or arthralgia). Each of these manifestations appears to be present in at least 50% of lupus patients at the time of diagnosis. The other clinical features of SLE are much less likely to be presenting manifestations, although virtually any of them might be the first clue to the correct diagnosis. More commonly, these manifestations appear over time as the disease evolves. Taken together, various descriptive studies in the literature1,8–12 support a cumulative frequency of symptoms and signs that is summarized in Table 80-2.
Table 80-2 Frequencies of Various Manifestations of Systemic Lupus Erythematosus*
| Manifestation | Frequency |
|---|---|
| Constitutional symptoms (fatigue, fever, weight loss) | 90%-95% |
| Mucocutaneous involvement (malar rash, alopecia, mucosal ulcers, discoid lesions, etc.) | 80%-90% |
| Musculoskeletal involvement (arthritis/arthralgia, avascular necrosis, myositis, etc.) | 80%-90% |
| Serositis (pleuritis, pericarditis, peritonitis) | 50%-70% |
| Glomerulonephritis | 40%-60% |
| Neuropsychiatric involvement (cognitive impairment, depression, psychosis, seizures, stroke, demyelinating syndromes, peripheral neuropathy, etc.) | 40%-60% |
| Autoimmune cytopenia (anemia, thrombocytopenia) | 20%-30% |
* Systemic lupus erythematosus is a heterogeneous disease that can affect virtually any organ system in variable ways.
Mucocutaneous Involvement
Mucocutaneous involvement is very common in SLE. Gilliam and colleagues have categorized cutaneous lupus erythematosus (LE) lesions as “lupus specific” or “lupus nonspecific” based on the histopathologic finding of interface dermatitis13,14 (Table 80-3). The presence of lupus-specific lesions confirms the diagnosis of cutaneous LE; lupus nonspecific lesions can occur in diseases other than lupus. Lupus-specific lesions are further subdivided into acute lupus erythematosus (ACLE), subacute lupus erythematosus (SCLE), and chronic cutaneous lupus erythematosus (CCLE) lesions, based on additional clinical and histopathologic information. Discoid lupus is the most common subtype of CCLE. SCLE and CCLE can occur as distinct isolated entities or as one of several manifestations of SLE. The risk of SLE varies with each cutaneous subset. One study of 161 patients with lupus-specific lesions showed that the classification criteria for SLE were present in 72% of patients with ACLE, 58% with SCLE, 28% with any form of discoid lupus, and 6% with localized discoid lupus confined to head and neck. Patients commonly displayed more than one type of cutaneous lesion.15
Table 80-3 Gilliam Classification of Skin Lesions Associated with Lupus
Acute Cutaneous Lupus Erythematosus
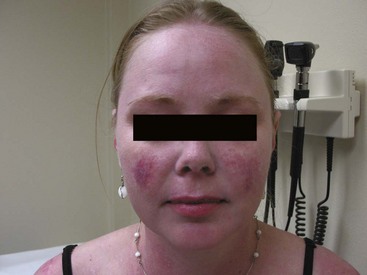
Acute cutaneous lupus erythematosus (ACLE) lesions can be localized or generalized. The hallmark feature of ACLE is localized to the malar region (“butterfly rash”) and is characterized by confluent, macular or papular erythema lasting days to weeks that occurs symmetrically on the cheeks and bridge of the nose, sparing the nasolabial folds (Figure 80-1). Induration and scaling may occur. The malar rash of SLE can be mimicked by a variety of other facial rashes, including acne, rosacea, seborrheic dermatitis, perioral dermatitis, atopic dermatitis, and erysipelas. If the diagnosis remains uncertain after an extensive clinical and serologic evaluation, biopsy of the rash can aid in distinguishing cutaneous lupus from other dermatologic conditions. It is important to remember that other forms of lupus-specific skin lesions such as discoid lupus can also occur in the malar distribution.
Subacute Cutaneous Lupus Erythematosus
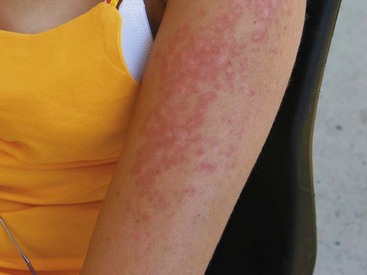
Subacute cutaneous lupus erythematosus (SCLE) is characterized by the presence of nonscarring, photosensitive lesions that can take one of two distinct forms: (1) papulosquamous lesions that resemble psoriasis, or (2) annular-polycyclic lesions with peripheral scale and central clearing (Figure 80-2). These two forms can occur concurrently in the same patient. SCLE has a predilection for the back, neck, shoulders, and extensor surfaces of the arms and usually spares the face. The lesions typically last for weeks to months and heal without scarring. Uncommonly, a severe TEN-like eruption can evolve from SCLE lesions after sun exposure.16 SCLE, particularly the annular subtype, is strongly associated with the presence of anti-SSA/Ro antibody.17 Several drugs are known to induce SCLE; angiotensin-converting enzyme inhibitors, terbinafine, hydrochlorothiazide, and calcium channel blockers are common culprits. Finally, SCLE has been implicated as a paraneoplastic syndrome.18
Chronic Cutaneous Lupus Erythematosus
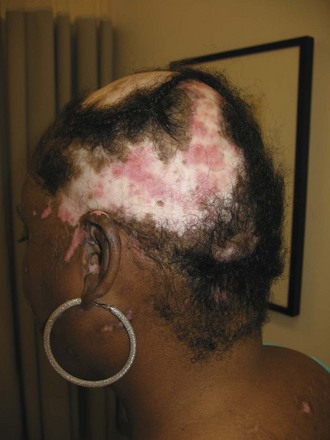
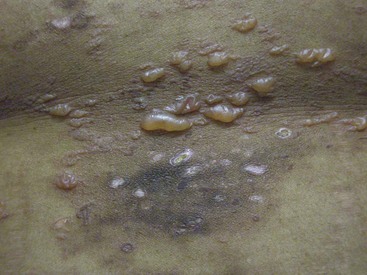
Chronic cutaneous lupus erythematosus (CCLE) refers to a variety of subtypes of photosensitive lesions that can lead to skin atrophy and scar and that may persist for several months. Discoid lupus (DLE) is the most common subtype of CCLE and is subdivided into localized discoid lupus (limited to head and neck) and generalized discoid (occurring above and below the neck) (Figures 80-3 and 80-4). The term “discoid” refers to the sharply demarcated disk-shaped appearance of the lesions. The lesions are raised, erythematosus plaques with adherent scale that commonly occur on the scalp, face, and neck. The cheeks, nose, ears, and upper lip are classic locations. Typically, a raised, erythematous border denotes the actively expanding component. Follicular plugging is a characteristic finding. Left untreated, DLE can result in permanent alopecia and disfigurement. Squamous cell carcinoma has been described as a sequela of long-standing DLE; thus, active surveillance of known lesions and evaluation of changing lesions are critical.19 Other subtypes of DLE include mucosal DLE (described later) and hypertrophic LE. Hypertrophic LE consists of chronic, indurated lesions that are covered by hyperkeratotic, multilayered scales. These lesions can be a source of diagnostic confusion because they may visually and histologically resemble squamous cell carcinoma.20
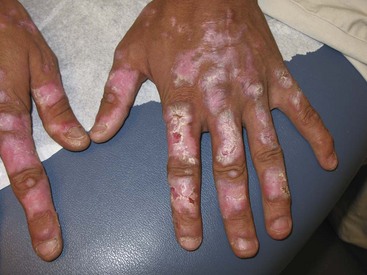
Other forms of CCLE include lupus panniculitis/profundus and chilblain lupus. Lupus panniculitis is a lobular panniculitis that has a predilection for the scalp, face, arms, buttocks, and thighs. When a cutaneous discoid lesion overlies the panniculitis, the entity is referred to as lupus profundus.21 Biopsy is often necessary to secure the diagnosis because reports have described T cell lymphoma mimicking panniculitis. However, biopsy should be performed carefully because the lesions have a tendency to break down. Lupus panniculitis is one of the few panniculitides that can occur above the waist. Lupus panniculitis is associated with low risk of concomitant SLE. Chilblain lupus manifests as tender, erythematous, or violaceous papules occurring on acral areas, especially fingers, toes, heels, nose, and ears. The lesions are brought on by cold, damp air.
Other Systemic Lupus Erythematosus Skin Lesions
SLE patients can develop lupus-nonspecific skin lesions such as cutaneous leukocytoclastic vasculitis, bullous lesions, periungal erythema, and livedo reticularis. Cutaneous leukocytoclastic vasculitis most commonly presents as palpable purpura on the lower extremities. Bullous lupus erythematosus is a rare cutaneous manifestation characterized by subepidermal vesiculobullous skin changes. It is manifested by a nonscarring bullous eruption22 (Figure 80-5). SLE may be associated with other bullous disorders such as bullous pemphigoid and dermatitis herpetiformis. The physical examination finding of periungal erythema represents dilation of the capillaries at the base of the nail. These capillaries can be visualized at the bedside with a dermatoscope or ophthalmoscope. Other disorders associated with periungal erythema include scleroderma and mixed connective tissue disease (MCTD). Unlike scleroderma and MCTD, SLE is not associated with capillary dropout. Livedo reticularis is characterized by an erythematous to violaceous reticular or net-like pattern of the skin. It is highly associated with the antiphospholipid antibody syndrome.
Photosensitivity
Photosensitivity occurs frequently in SLE. In one study, photoprovocation testing caused an abnormal skin reaction to ultraviolet A, ultraviolet B, or visible light in greater than 90% of lupus patients.23 Most abnormal skin reactions occurred 1 to 2 weeks after exposure to light and persisted for weeks to months. Photosensitive patients may report worsening of their systemic disease symptoms such as fatigue and joint pain following sun exposure. During evaluation of a photosensitive patient, polymorphous light eruption (PMLE) and phototoxic medications are important diagnostic considerations.24 Accurate differentiation between PMLE and lupus is essential because PMLE is treated by ultraviolet radiation phototherapy, but lupus is worsened by it. PMLE can occur concomitantly in patients with known SLE.
Alopecia
Scarring alopecia is a common complication of discoid lupus. Scalp discoid lesions most frequently develop on the vertex and parietal areas.25 Nonscarring alopecia in SLE patients can take several forms. “Lupus hair” is characterized by short, irregularly sized hair at the frontal hairline and is associated with active systemic disease.26 Telogen effluvium manifests as diffuse hair thinning. Lastly, the incidence of alopecia areata (discrete areas of hair loss) is believed to be increased in SLE.27
Mucosal Ulcers
SLE patients commonly develop nasal or oral lesions that represent the mucosal counterparts of cutaneous lupus.28 Acute oral lupus lesions present as red macules, palatal erythema or petechiae, erosions, or ulcerations. These lesions are usually painless. Subacute oral lesions are rare, and are characterized by well-demaracted, round, red patches. Oral discoid lesions present as painful, well-demarcated, round, red lesions with white radiating hyperkeratotic striae. When the lesions evolve, they may take on a honeycomb appearance. Oral discoid lupus frequently involves the lip and spreads from the vermilion border to the skin of the lip. Lupus oral ulcers have a gradual onset and can occur anywhere on the oral mucosa, the most common locations being the hard palate, buccal musosa, and vermilion border.29 These lesions are most commonly unilateral or asymmetric. The relationship between the presence of oral lesions and systemic disease activity remains unclear. Note that oral candidiasis and oral lichen planus can take on a similar appearance to SLE oral ulcers. The histopathology and immunopathology of musosal lesions are similar to the alterations seen in the skin. Vasculitis is absent.
Dermatopathology and Immunopathology
A skin biopsy is useful in the diagnosis of cutaneous lupus in the setting of an atypical clinical presentation. Immunofluorescence should always be performed along with conventional histology. “Lupus-specific” skin lesions are characterized by an interface dermatitis consisting of a mononuclear cell infiltrate at the dermal-epidermal junction. Other pathologic findings present in lupus skin lesions include basal layer vasculopathic degeneration of keratinocytes, perivascular and periadnexal inflammation, follicular plugging, mucin deposition, and hyperkeratosis. These findings occur to different degrees in various lupus-specific skin lesions, with discoid lesions showing the most profound changes.30 In contrast, early ACLE lesions may have minimal histopathologic findings and only a sparse lymphocytic infiltrate.
Immunofluorescence demonstrates granular deposition of immunoglobulin and complement components along the dermal-epidermal junction. Immunoglobulin (Ig)G and IgM are the most common immunoglobulin subtypes deposited. Various complement components including C3, C1q, and the membrane attack complex have also been identified. Direct immunofluorescence (DIF) of nonlesional, sun-exposed skin is called the lupus band test. Although a positive lupus band test is often seen in patients with SLE, this may also be seen in patients with other rheumatic diseases, as well as in healthy people. In one study, 20% of healthy young adults had a positive DIF in sun-exposed skin.31 It is important to note that serologic testing with antinuclear antibody (ANA), anti–double-stranded DNA (anti-dsDNA), and anti-Smith (anti-SM) has largely supplanted use of the lupus band test in confirming the diagnosis of SLE. DIF of nonlesional, sun-protected skin is believed to be more specific for SLE.
Musculoskeletal
Arthritis
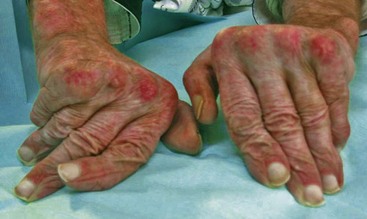
Arthritis and arthralgias are very common manifestations of SLE, present in up to 90% of patients at some point during the course of their disease.32 Severity of involvement can range from mild joint pain to deforming arthritis. Although any joint can be involved, lupus arthritis is characterized by a symmetric, inflammatory arthritis predominantly affecting the knees, wrists, and small joints of the hands.33 Synovial effusions are typically small and not as inflammatory as those present in rheumatoid arthritis. Hand deformities can occur as a result of ligamental and/or joint capsule laxity and joint subluxation. This manifestation is called “Jaccoud’s-like arthropathy” because it resembles the arthropathy that develops in patients with a history of rheumatic fever (Figure 80-6). Lupus hand deformities are reducible. Jaccoud’s-like arthropathy sometimes occurs in the foot as well.34
Although lupus arthritis is not classically associated with erosions on plain radiography, erosive disease has been described in a small subset of patients.35 In addition, MRI studies have shown occasional erosions in some patients with lupus arthritis.36 Erosive arthritis is more commonly a feature of MCTD. Some studies have demonstrated an association between lupus erosive arthritis and the presence of anticyclic citrullinated protein (anti-CCP) antibodies.35 In addition to arthritis, tendinitis or tenosynovitis is frequently observed in SLE patients.37 Tendon rupture is a very uncommon occurrence.
Synovial biopsies from lupus arthritis patients have shown a variety of abnormalities, including deposition of fibrin-like material, focal or diffuse synovial lining cell proliferation, vascular congestion, perivascular mononuclear cell infiltration, vasculitis, and obliteration of vessel lumina.38 Radiographic studies of patients with SLE have revealed changes such as cystic bone lesions, periarticular soft tissue swelling, demineralization, acral sclerosis, joint subluxations, and erosions.39,40
Avascular Necrosis
Avascular necrosis (AVN), also referred to as aseptic necrosis and ischemic necrosis, is a painful and disabling condition that occurs in some SLE patients.41 AVN is the end result of interruption of the blood supply to bone, leading to reactive hyperemia of adjacent bone, demineralization, and then collapse. The most commonly affected sites include the femoral heads, tibial plateaus, and femoral condyles, but smaller joints can be involved as well. AVN is often bilateral, and joint effusions may occur. AVN of the femoral head should be suspected in an SLE patient with groin pain that is worsened with weight bearing and movement of the hip. The pain may radiate down the side of the thigh, and a limp might be evident. Although both plain radiographs and magnetic resonance imaging (MRI) can be helpful in the diagnosis of AVN, MRI is the more sensitive test. One prospective MRI study of 45 SLE patients on glucocorticoid therapy demonstrated that 34% of patients developed silent osteonecrosis of the femoral head.42 However, MRI studies may be too sensitive an indicator, in that some lupus patients with suggestive MRI findings never progress to clinical symptoms of AVN. Therefore, MRI findings should always be interpreted in the context of the clinical setting. The use of high doses of glucocorticoids is a well-known risk factor for the development of AVN, but AVN has also been described in SLE patients who have never used glucocorticoids. An MRI study of 72 newly diagnosed SLE patients demonstrated that AVN typically developed within the first 3 months of initiation of high-dose glucocorticoids.43 Epidemiologic studies have shown that high disease activity as measured by the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) and the use of cytotoxic medications are also associated with AVN.44,45
Myositis
Although myalgias occur commonly in SLE, true myositis is relatively rare. One study of SLE patients at the National Institutes of Health (NIH) found a prevalence of myositis of 8%.46 In most of those patients, myositis was one of the presenting features of SLE. Myositis usually involves the proximal upper and lower extremities. Histologic findings in SLE myositis are often less pronounced than those observed in polymyositis.
A biopsy study of 55 unselected SLE patients demonstrated that several pathologic changes, including type II fiber atrophy, lymphocytic vasculitis, and myositis, were increased in SLE patients compared with control patients.47
Renal Involvement
General Considerations
Renal involvement is common in SLE and is a significant cause of morbidity and mortality.48 It is estimated that up to 90% of SLE patients will have pathologic evidence of renal involvement on biopsy, but only 50% will develop clinically significant nephritis. The clinical presentation of lupus nephritis is highly variable, ranging from asymptomatic hematuria and/or proteinuria to frank nephrotic syndrome to rapidly progressive glomerulonephritis with loss of renal function. Lupus nephritis typically develops within the first 36 months of the disease, although there are exceptions. Thus, periodic screening for the presence of nephritis is a critical component of the ongoing evaluation and management of SLE patients. Routine screening procedures include inquiring about new-onset polyuria, nocturia, or foamy urine and looking for the presence of hypertension or lower extremity edema. It is important to screen at regular intervals for the presence of proteinuria and/or hematuria and a change in serum creatinine; in active SLE patients, screening at 3-month intervals is prudent.
Types of Renal Involvement in Systemic Lupus Erythematosus
Several forms of renal involvement have been noted in SLE, including immune complex–mediated glomerulonephritis (most common form), tubulointerstitial disease, and vascular disease. Glomerulonephritis is characterized by immune complex deposition and inflammatory cell infiltration into the glomerulus. The pattern of glomerular injury is primarily related to the site of immune complex deposition. Tubulointerstitial and vascular disease can occur with or without immune complex–mediated glomerulonephritis. Tubulointerstitial disease has been observed in up to 66% of SLE renal biopsy specimens49 and is characterized by inflammatory cell infiltrates, tubular damage, and interstitial fibrosis. The presence of tubulointerstitial disease is a strong predictor of poor long-term renal outcome.50
Renal vascular lesions in SLE include “lupus vasculopathy,” thrombotic microangiopathy (TMA), vasculitis, and nonspecific vascular sclerosis.51,52 Lupus vasculopathy is defined as the presence of immunoglobulin and complement-containing hyaline thrombi within the glomerular capillary or arteriolar lumina. Inflammatory changes to the vascular wall are absent. TMA is characterized by the presence of fibrin thrombi within the glomerular capillary or arteriolar lumina and may be associated with the presence of antiphospholipid antibodies. The finding of TMA should prompt consideration of antiphospholipid antibody syndrome nephropathy (APSN). Although exceedingly rare, true vasculitis characterized by leukocyte infiltration and fibrinoid necrosis of arterial walls can occur. Nonspecific sclerotic vascular lesions characterized by fibrous intimal thickening are commonly observed. The presence of such vascular lesions is associated with decreased renal survival.53 In addition to the lupus-related renal lesions described previously, SLE patients may develop renal abnormalities that are unrelated to their underlying SLE. Such pathologic lesions include focal segmental glomerulosclerosis (FSGS), hypertensive nephrosclerosis, and thin basement membrane disease.54 In an SLE patient in whom renal disease is suspected, renal biopsy is critical in distinguishing between these potential causes and in guiding appropriate management decisions.
Laboratory Evaluation
Urinalysis
Performance of a urinalysis with microscopy is essential in the screening and monitoring of lupus nephritis.55 Hematuria, pyuria, dysmorphic red blood cells, red blood cell casts, and white blood cell casts may all be present. Red blood cell casts are very specific, but not sensitive, for the diagnosis of glomerulonephritis. Early morning urine specimens, which tend to be concentrated and acidic, are ideal for the detection of red blood cell casts. White blood cells, red blood cells, and white blood cell casts may indicate the presence of tubulointerstitial involvement. Hematuria in the absence of proteinuria might be due to urolithiasis, menstrual contamination, or bladder pathology, particularly transitional cell carcinoma in a patient with previous cyclophosphamide exposure.
Accurate measurement of proteinuria is critical because proteinuria is a very sensitive indicator of glomerular damage. In addition, studies of chronic kidney disease have shown that the magnitude of proteinuria is a strong predictor of glomerular filtration rate decline.56 Normal daily protein excretion is less than 150 mg. Although the gold standard tool is an accurately collected 24-hour urine protein, this test can be cumbersome for patients and is prone to errors in undercollection and overcollection. Thus, many clinicians are currently using the random spot urine protein-to-creatinine ratio out of convenience. Use of the spot ratio is controversial because data suggest that the spot ratio often is not representative of the findings in a timed collection, especially in the range of 0.5 to 3.0 (the range of most lupus nephritis flares).57 However, a spot ratio can be a helpful screening test for the presence of proteinuria and is useful in differentiating nephrotic from non-nephrotic range proteinuria.58 Urine dipstick should not be used for the quantification of proteinuria because it reflects protein concentrations and varies depending on the volume of the sample. Many experts currently recommend calculation of the protein:creatinine ratio from a 12- or 24- hour urine collection as the gold standard of proteinuria assessment.59
Renal Biopsy
Before renal biopsy, ultrasonography is recommended to assess kidney size and structure and to rule out renal vein thrombosis. Kidney size of less than 75% of normal is a relative contraindication to biopsy.60 SLE glomerulonephritis is classified by the International Society of Nephrology/Renal Pathology Society (ISN/RPS) into six categories based on light microscopic, immunofluorescent, and electron micrographic findings61 (Table 80-4 and Figure 80-7).
Table 80-4 International Society of Nephrology/Renal Pathology Society Classification of Lupus Nephritis
| WHO Type | |
|---|---|
| Class I | Minimal Mesangial Lupus Nephritis |
| Normal glomeruli by light microscopy, but mesangial immune deposits by immunofluorescence | |
| Class II | Mesangial Proliferative Nephritis |
| Purely mesangial hypercellularity of any degree or mesangial matrix expansion by light microscopy, with mesangial immune deposits. Few isolated subepithelial or subendothelial deposits may be visible by immunofluorescence or electron microscopy, but not by light microscopy | |
| Class III | Focal Lupus Nephritis |
| Active or inactive focal, segmental, or global endocapillary or extracapillary glomerulonephritis involving <50% of all glomeruli, typically with focal subendothelial immune deposits, with or without mesangial alterations | |
| Class IV | Diffuse Lupus Nephritis |
| Active or inactive diffuse, segmental, or global endocapillary or extracapillary glomerulonephritis involving ≥50% of all glomeruli, typically with diffuse subendothelial immune deposits, with or without mesangial alterations. This class is subdivided into diffuse segmental (IV-S) lupus nephritis when ≥50% of the involved glomeruli have segmental lesions, and diffuse global (IV-G) lupus nephritis when ≥50% of the involved glomeruli have global lesions. Segmental is defined as a glomerular lesion that involves less than half of the glomerular tuft. This class includes cases with diffuse wire loop deposits, but with little or no glomerular proliferation | |
| Class V | Membranous Lupus Nephritis |
| Global or segmental subepithelial immune deposits or their morphologic sequelae by light microscopy and by immunofluorescence or electron microscopy, with or without mesangial alterations Class V nephritis may occur in combination with class III or class IV, in which case both are diagnosed Class V nephritis may show advanced sclerotic lesions | |
| Class VI | Advanced Sclerotic Lupus Nephritis |
| ≥90% of glomeruli globally sclerosed without residual activity |
WHO, World Health Organization.
Adapted from Weening JJ, et al: The classification of glomerulonephritis in systemic lupus erythematosus revisited, J Am Soc Nephrol 15:241, 2004.
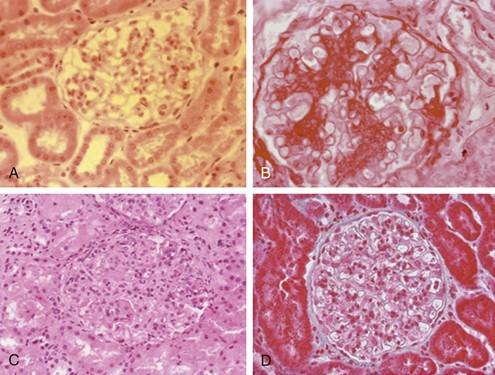
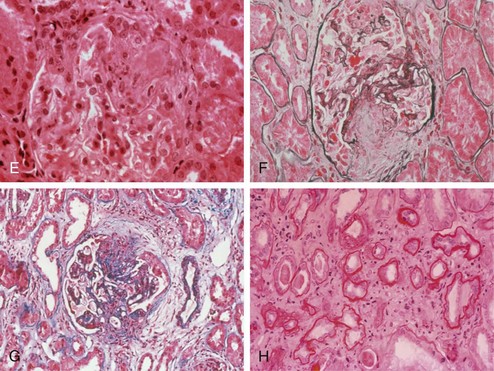
Figure 80-7 A through D, World Health Organization types of lupus. (See Table 80-4 for a detailed description of histologic findings.) A, Normal glomerulus (type I). B, Mesangial proliferative (type II). C, Proliferative nephritis. Dramatic increase in mesangial and endocapillary cellularity produces a lobular appearance of the glomerular tufts and compromises the patency of most capillary loops. When less than 50% of glomeruli are involved, nephritis is denoted as focal (type III). When more than 50% of glomeruli are involved, nephritis is denoted as diffuse (type IV). D, Membranous nephropathy (type V). In membranous lupus nephropathy, the capillary walls of the glomerular tuft are prominent and widely patent, resembling “stiff” structures with decreased compliance. E through H, High-risk histologic features suggesting severe nephritis. E, Fibrinoid necrosis with karyorrhexis in a patient with focal proliferative glomerulonephritis. F and G, Cellular crescents with layers of proliferative endothelial cells and monocytes lining Bowman’s capsule along with a predominantly mononuclear interstitial infiltrate. H, Severe interstitial fibrosis and tubular atrophy. Note the thickening of the tubular basement membranes and tubular epithelial degeneration with separation of residual tubules caused by deposition of collagenous connective tissue among tubules.
Renal biopsy is especially important because urinary parameters such as hematuria and the degree of proteinuria imperfectly predict the underlying renal pathology.62,63 Hematuria might be absent in patients with severe class IV nephritis, and proteinuria can be modest in patients with class V nephritis. A repeat renal biopsy may be indicated in certain clinical settings (e.g., if a patient is not responding appropriately to therapy, if a patient unexpectedly worsens after having achieved a good response to therapy). Repeat renal biopsy can be useful in detecting class transformation that occurs in 15% to 50% of lupus nephritis patients during the course of their disease. Class transformation can occur spontaneously or as a result of treatment.
Outcome
Each ISN/RPS histopathologic class portends a distinct renal prognosis. Patients with class I and class II nephritis have an excellent renal prognosis and do not require any specific therapy. In contrast, the long-term renal prognosis of class III or class IV nephritis is believed to be poor in the absence of immunosuppressive therapy. Although the long-term renal prognosis of class V nephritis is more favorable than that of class III or class IV nephritis, class V patients are more likely to suffer from morbid complications of the nephrotic syndrome, including cardiovascular disease, thromboembolic disease, and hyperlipidemia. Several epidemiologic studies have defined demographic, clinical, and histopathologic factors associated with renal outcome in patients with lupus nephritis. Studies have shown that African-Americans and Hispanics/Latinos generally experience a worse renal prognosis than white and Chinese populations. The reasons for this disparity most likely involve a combination of genetic and socioeconomic factors. A retrospective analysis of 65 patients at the NIH suggested that age greater than 30 years, African-American race, low hematocrit, elevated serum creatinine, and low C3 complement were associated with increased probability of renal failure. The histologic features of cellular crescents and interstitial fibrosis were also associated with worse renal prognosis.50
Pleuropulmonary Involvement
Pleuropulmonary manifestations of SLE are diverse and can involve any aspect of the lung (Table 80-5).
Table 80-5 Pleuropulmonary Manifestations of Systemic Lupus Erythematosus
| Manifestation | Key Features |
|---|---|
| Pleuritis | May occur with or without effusion May correlate with elevated serum C-reactive protein |
| Pleural effusion | May be asymptomatic Usually small, bilateral, exudative Common feature of drug-induced lupus Must exclude infection, malignancy, heart failure |
| Acute pneumonitis | Severe respiratory illness with fever, cough, pulmonary infiltrates, hypoxemia Pleural effusion may be present High mortality rate Bronchoscopy with bronchoalveolar lavage might be necessary to exclude infection |
| Chronic interstitial lung disease | May develop after acute pneumonitis or in a more insidious fashion Presents as dyspnea on exertion, pleuritic chest pain, nonproductive cough High-resolution computed tomography more sensitive than chest x-ray in detecting disease Must exclude infection, pulmonary edema, malignancy |
| Diffuse alveolar hemorrhage | Presents as dyspnea and cough, alveolar infiltrates, fall in blood hemoglobin level Hemoptysis may not be present Diffusion capacity of carbon monoxide typically increased Bronchoscopy with bronchoalveolar lavage confirms the diagnosis and excludes infection High mortality rate |
| Pulmonary arterial hypertension | Presents as dyspnea on exertion, fatigue, chest pain, nonproductive cough Diagnosis should be confirmed with right heart catheterization Exclude secondary causes of pulmonary hypertension, including thromboembolic disease |
| Shrinking lung syndrome | Dyspnea, low lung volumes, elevation of hemi-diaphragms in absence of lung parenchymal involvement |
Pleuritis
Up to 50% of SLE patients will develop pleuritis. Clinically apparent pleural effusions are typically small, bilateral, and exudative.64 Pleuritis is commonly manifested by pleuritic chest pain, but pleural effusions may be asymptomatic and detected on routine chest radiography performed for another purpose. Massive pleural effusions requiring pleurocentesis and/or pleurodesis are uncommon but have been reported.65 The presence of pleuritis usually corresponds to active SLE in other organ systems.66 Thoracoscopic evaluation has demonstrated nodules on the visceral pleura with immunoglobulin deposits detected on immunfluorescence. The differential diagnosis of pleural effusions in an SLE patient includes infection, malignancy, and heart failure. In addition, pleural effusions are a common feature of drug-induced lupus. In the absence of infection, high levels of serum C-reactive protein (CRP) have been found to correlate well with the presence of pleuritis and other forms of serositis in SLE.66,67 Thus, serum CRP may be a useful clue to the presence of pleuritis.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


