64 Cell-Targeted Biologics and Targets
Rituximab, Abatacept, and Other Biologics
As appreciation of the gravity of the social and economic burden imposed by rheumatoid arthritis (RA) has grown, so has the recognition that more favorable clinical outcomes are achieved when synovitis is optimally suppressed. The evidence is particularly compelling early in the course of RA, when intervention with disease-modifying combination therapy results in improved remission rates and increased clinical and radiographic benefits.1–3 The armamentarium of potential therapeutics has also grown with the identification of relevant disease molecules. Of these, biologic therapeutics targeting tumor necrosis factor (TNF), particularly when used in combination with oral methotrexate, have enjoyed notable success in suppressing inflammation and markedly inhibiting the progression of structural damage previously thought to be an unavoidable characteristic of RA.4,5 However, despite the unprecedented clinical and commercial successes of TNF inhibitors, their availability is restricted by high costs. In addition, a substantial proportion of RA patients fail to demonstrate significant clinical responses.
Targeting B Cells
B cells arise from stem cells in the bone marrow, where they acquire an antibody receptor bearing a unique variable region. A number of maturation and activation steps take place as the B cells migrate from the marrow compartment, through blood, and to perifollicular germinal centers and memory compartments in lymphoid tissue, before returning to the marrow as mature plasma cells.6 Successful maturation and survival of cells are tightly regulated and dependent on a number of trophic signals delivered via cell surface ligands such as vascular cell adhesion molecule-1 (VCAM-1) and soluble factors such as B lymphocyte stimulator (BLyS).7,8
In the late 1990s, Edwards and colleagues9,10 suggested that the (assumed) underlying autoreactive response in RA might be driven by self-perpetuating B cells and that the initiation of inflammation results from ligation of the low-affinity immunoglobulin G (IgG) receptor FcRγIIIa by immune complexes. An attractive feature of this hypothesis, particularly in seropositive patients, is that it might account for the tissue tropisms of disease expression in the RA syndrome complex because FcRγIIIa is expressed in high levels in synovium and other extra-articular tissues that may be involved in RA. RF-producing cells can capture antibodies bound to antigen before antigen internalization by endocytosis and subsequent presentation of peptide fragments to a T cell, with provision of T cell help to the B cell. Edwards and colleagues11 also proposed that such RF-producing B cells might become self-perpetuating by an amplification signal arising from co-ligation of the B cell receptor and small immune complexes formed by IgG RF bound to the complement component C3d, providing a survival signal. In contrast, co-ligation of certain other B cell surface receptors with the B cell receptor may provide a negative survival signal. In the rare event that self-perpetuating, autoreactive B cells arise, having escaped normal regulatory mechanisms, this theory predicts that a B cell depletion strategy would remove the autoreactive B cell clones and their antibody products. Because CD20 is not internalized and is highly expressed on a range of B lineage cells including pre-B cells, immature B cells, activated cells, and memory cells, but is not found on stem, dendritic, or plasma cells (Figure 64-1), it is an ideal target for B cell depletion by monoclonal antibodies. The hypothesis that B cells represent a therapeutic target in RA is also being tested in the clinic using other strategies for B cell inhibition, as discussed later.
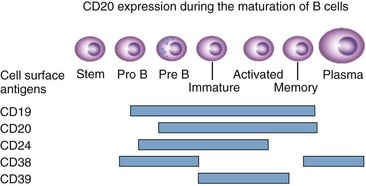
The CD20 antigen is located in the B cell membrane, with 44 amino acids exposed to the extracellular space. Its function is unknown, although it may have a role in cell signaling or in calcium mobilization.12 Interestingly, CD20 knockout mice do not have a clear-cut phenotype or obvious B cell defect.13 CD20+ B cells represent a prominent population in the rheumatoid synovial tissue in the majority of patients.
Rituximab in Rheumatoid Arthritis
Key Points
Rituximab is an effective biologic therapy across the spectrum of RA-patient populations.
It appears to have greatest benefit in seropositive patients.
Rituximab given as two infusions of 1 g each slows radiographic progression in RA.
Rituximab is a chimeric mouse-human monoclonal antibody directed against the extracellular domain of the CD20 antigen. It initiates complement-mediated B cell lysis and may permit antibody-dependent, cell-mediated cytotoxicity when the Fc portion of the antibody is recognized by corresponding receptors on cytotoxic cells. Rituximab may also initiate apoptosis14 and influence the ability of B cells to respond to antigen or other stimuli.15 Rituximab initially found a role in the clinic as a single-agent treatment for relapsed or refractory low-grade or follicular CD20+ B cell non-Hodgkin’s lymphoma, for which it was approved. For this reason, there was a wide experience with rituximab in hematologic oncology before clinical trials in RA and its recent approval in the United States and Europe for the treatment of TNF inhibitor–refractory RA patients with active disease.
Following rituximab administration, rapid B cell depletion takes place in peripheral blood and conventional methods for measurement of peripheral blood B cells by means of CD19 expression detect no cells at all in the majority of cases. Investigation of synovial tissue from patients with RA treated with rituximab reveals a decrease in synovial B cells and plasma cells in most but not all patients.16,17 These findings raise the possibility that there might be as yet poorly understood rescue mechanisms in the inflamed synovium providing survival niches or, alternatively, that some B cells may have an inherent resistance against depletion.
Analysis of peripheral blood memory B cells before depletion and during and after reconstitution seems to be predictive of clinical outcome, with patients showing early relapses having substantially higher IgD+ and IgD-CD27+ memory B cell numbers and proportion during B cell recovery.18 In addition to these cellular biomarkers, serologic parameters have been analyzed. Decreases in RF or anticitrullinated protein antibody serum levels are reported to be associated with B cell depletion,19 but further studies are necessary to determine the relationships between these serologic changes and clinical response.
Clinical Studies
The findings of early clinical studies of B cell depletion therapy in patients with active RA using rituximab in a number of different treatment regimens suggested an encouraging benefit with an acceptable safety profile and pointed to a possible therapeutic role for rituximab in RA.20–22 Confirmation of benefit, however, required a randomized, double-blind, controlled study. In a phase IIa study, the efficacy of rituximab in active RA was tested in 161 patients who had failed to respond adequately to methotrexate at a dose of at least 10 mg a week for a minimum of 16 weeks.23 Patients were assigned to one of four treatment regimens: 1-g infusion of intravenous rituximab alone on days 1 and 15, methotrexate alone as a comparison arm, intravenous rituximab with cyclophosphamide infusions at a dose of 750 mg on days 3 and 17, or rituximab and methotrexate. All patients received 100 mg methylprednisolone just before each treatment, in addition to prednisolone 60 mg daily on days 2, 4, 5, 6, and 7 and 30 mg daily on days 8 to 14. The primary end point was the proportion of patients achieving an American College of Rheumatology 50% improvement criteria (ACR50) response at week 24, and exploratory analyses were undertaken at week 48. At week 24, a significantly greater proportion of patients achieved an ACR50 in the rituximab and methotrexate combination group (43%; P = .005) and in the rituximab and cyclophosphamide combination group (41%; P = .005) than in the group receiving methotrexate as monotherapy (13%) (Table 64-1). Thirty-three percent of the patients receiving rituximab alone achieved an ACR50 response, but this failed to reach statistical significance compared with methotrexate alone (P = .059). In all the rituximab groups, the mean change from baseline in disease activity score was significant compared with methotrexate alone.

Rituximab treatment was associated with near-complete peripheral blood B cell depletion, persisting throughout the 24-week period of the primary analysis. Patients in the rituximab groups had a substantial and rapid reduction in the concentration of RF in serum, but despite peripheral B cell depletion, immunoglobulin levels did not change substantially.23 The overall incidence of infection was similar in the control and rituximab groups at 24 and 48 weeks. By week 24, four patients in the rituximab groups had suffered a serious infection, as well as one in the control group. Two additional serious infections were reported during the extended 48-week period in the rituximab groups, one of which was fatal. Infusion reactions of any type were reported in 36% of patients receiving rituximab and 30% of patients receiving placebo, although most were characterized as mild or moderate. The reactions included hypotension, hypertension, flushing, pruritus, and rash. In the rare case of severe reactions, it has been suggested that a cytokine release syndrome associated with marked cell lysis following rituximab might be a contributing factor.
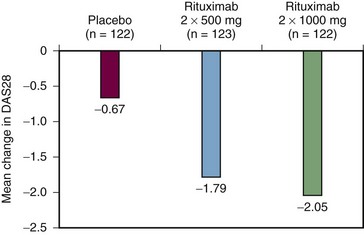
To follow up the phase IIa study, a phase IIb study was undertaken to examine the efficacy and safety of rituximab at different doses, with or without glucocorticorticoids, in patients with active RA resistant to DMARDs including biologics. The findings of this phase IIb study, known as the DANCER (Dose-ranging Assessment: International Clinical Evaluation of Rituximab in RA) trial, were reported in 2006.24 The researchers recruited 465 patients with active disease who had to have failed to respond to at least one DMARD other than methotrexate, but no more than five, or failed to respond to biologic response modifiers, and they had to have been treated with methotrexate as a single DMARD for at least 12 weeks, with 4 weeks of stable therapy at a dose of at least 10 mg a week. All other DMARDs were withdrawn at least 4 weeks before randomization—8 weeks for infliximab, adalimumab, and leflunomide. Patients were randomized to receive either placebo infusions or rituximab at a dose of 500 mg or 1 g on days 1 and 15, together with one of three glucocorticoid options: glucocorticoid placebo, 100 mg of intravenous methylprednisolone before each rituximab infusion, or 100 mg of methylprednisolone before each infusion in addition to an oral corticosteroid. The results at 24 weeks confirmed the significant efficacy of a single course of rituximab in active RA when combined with methotrexate. This benefit was independent of glucocorticoids, although methylprednisolone on day 1 reduced the incidence and severity of first rituximab infusion reactions by about one-third (Figure 64-2). Both rituximab doses were efficacious. At the lower dose, 55% of recipients achieved ACR20 responses, as did 54% of those at the higher rituximab dose—in both cases, significantly greater than the 28% of those receiving placebo infusions. Similarly, significantly higher proportions of patients achieved ACR50, ACR70 (see Table 64-1), and European League Against Rheumatism (EULAR) good responses at 24 weeks at both rituximab doses compared with patients receiving placebo infusions (Figure 64-3). At the most stringent ACR70 response level, the difference in the percentage of responders in the placebo, lower-dose rituximab, and higher-dose rituximab groups was most marked at the higher rituximab dose of 1 g 2 weeks apart (5%, 13%, and 20%, respectively; P < .05). Adverse events reported up to 24 weeks were largely infusion related, particularly at the time of the first infusion.
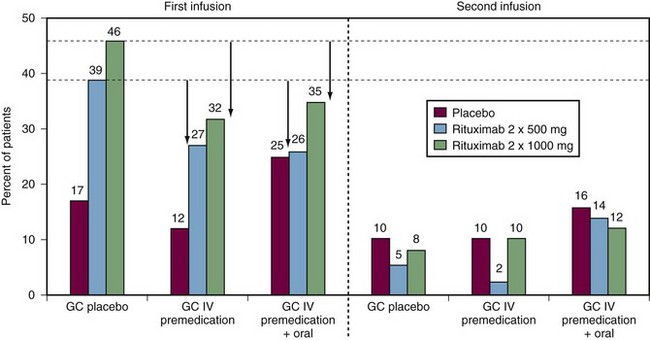
A trial, known by the acronym REFLEX (Randomized Evaluation of Long-term Efficacy of Rituximab in RA), was designed to determine the efficacy and safety of rituximab when used in combination with methotrexate in patients with active RA who have an inadequate response to one or more anti-TNF therapies because of either lack of efficacy (90% of patients recruited) or toxicity (10% of patients recruited); in addition, all patients had radiographic evidence of at least one joint with definite erosion attributable to RA (Figure 64-4). The recruited cohort comprised 520 patients with mean disease duration of 12 years on a background regimen of methotrexate 10 to 25 mg once a week. After a washout period during which other DMARDs and anti-TNF drugs were withdrawn, patients were randomized to receive a single course of 1 g rituximab or placebo infusions on days 1 and 15. All patients were given 100 mg intravenous methylprednisolone before each infusion and a brief course of oral prednisolone between the two doses: 60 mg daily from days 2 to 7 and 30 mg daily from days 8 to 14.25
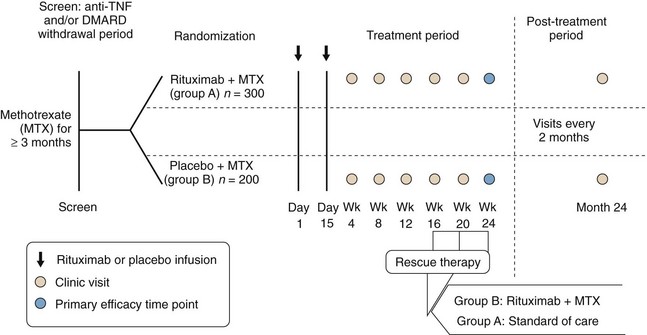
Of the patients assigned to rituximab, 82% completed 6 months, compared with only 54% of the patients assigned to placebo. The major reason for study withdrawal was lack of response, reported in 40% of the placebo group and 12% in the rituximab group. At 6 months, significantly more patients receiving rituximab achieved ACR20, ACR50, and ACR70 responses than did those receiving placebo: 51%, 27%, and 12%, respectively, on rituximab, versus 18%, 5%, and 1% of those on placebo (see Table 64-1). In terms of change in disease activity score (DAS28), intention-to-treat analyses showed that in patients administered placebo infusions, the reduction from baseline was 0.34, less than the 0.6-point reduction considered to be clinically meaningful; in contrast, the reduction was 1.83 in the rituximab group.25
The ACR response evaluates RA treatment on the basis of a 20%, 50%, or 70% improvement in five of seven core components. However, from the patient’s perspective, determining the actual benefit of an ACR20 improvement is not straightforward. In the REFLEX study, there were significantly greater improvements in all components of the ACR core measures in the rituximab group. Rituximab demonstrated a clinically meaningful benefit for RA patients in physical function evaluated by a health assessment questionnaire (HAQ) in all nonoverlapping ACR response categories. In the active treatment arm, both clinical and subjective parameters of the ACR core components contributed to the assignment of an ACR20 response, whereas in the placebo group, the subjective parameters dominated.25
In the REFLEX study, following a single treatment course, the maximal clinical response to rituximab plus methotrexate was observed at 24 weeks. After this time, patients were eligible to exit the study and receive further rituximab treatment on the basis of clinical need. Of the patients in the rituximab plus methotrexate group, 37% (114 of 308) remained in the study over 48 weeks, indicating continued clinical benefit after the single initial treatment course. The majority of patients who withdrew did so to receive further courses of rituximab between weeks 24 and 48 of the study. In contrast, 89% of the placebo plus methotrexate group (185 of 209) withdrew before week 48.26
Another recently reported phase III study known by the acronym SERENE (Study Evaluating Rituximab’s Efficacy in methotrexate iNadequate rEsponders) confirmed the benefits of rituximab in RA patients receiving concomitant methotrexate who had active disease at baseline despite methotrexate therapy and who had not received prior biologic therapy.27 Patients were randomized to receive either placebo or rituximab at one of two doses, 2 × 500 mg and 2 × 1000 mg. From week 24, in an open-label extension, those patients in the rituximab arms failing to achieve remission as assessed by DAS28 received a second course of rituximab and all placebo patients were treated with the lower rituximab dose. At week 24, a significantly greater proportion of patients receiving rituximab 2 × 500 mg or 2 × 1000 mg + methotrexate achieved the primary end point of an ACR20 response versus patients receiving placebo plus methotrexate (54.5% and 50.6% vs. 23.3%, respectively; P < .0001). By week 48 approximately 90% of patients in all treatment groups had received a second course of treatment. The majority of these repeat treatments (82% to 88%) were given by week 30.
Results from the IMAGE study, which included 748 patients without prior use of either biologics or methotrexate, were recently reported. In this group, high-risk patients who had baseline high DAS28 scores or high CRP were found to have greater DAS28 improvement at week 52 if they received rituximab in addition to methotrexate.28
Disease Modification
The extraordinary success of TNF blockade in inhibiting structural damage to joints in patients with RA has set a new standard to which all new biologics must aspire. Inhibition of structural joint damage by rituximab in patients with RA and a previous inadequate response to TNF inhibitors from the REFLEX study was first described over a 1-year period29 with a subsequent demonstration that the initial effects of rituximab are maintained over an extended interval of 2 years, with all measures of joint damage significantly improved compared with placebo plus methotrexate.30 At week 56,29 the mean change in Genant-modified Sharp score in the placebo plus methotrexate arm was 2.31, compared with 1.0 in the rituximab plus methotrexate group (P = .0043). Significant differences were also reported for joint space narrowing and bone erosions. At week 104, significantly lower changes in total Genant-modified Sharp score (1.14 vs. 2.81; P < .0001), erosion score (0.72 vs. 1.80; P < .0001), and joint space narrowing scores (0.42 vs. 1.00; P < .0009) were observed with rituximab plus methotrexate versus placebo plus methotrexate.30 Importantly, within the rituximab group, 87% who had no progression of joint damage at 1 year remained nonprogressive at 2 years. Thus these data confirm that rituximab plus methotrexate has the benefit of sustained inhibition of joint damage progression in patients with RA and a previously inadequate response to TNF inhibitors.
Radiographic outcomes were recently reported for a phase III study designed to determine the efficacy of rituximab in the prevention of joint damage and its safety in combination with methotrexate in the context of patients initiating treatment with methotrexate.28 This study, known by the acronym IMAGE (International study in Methotrexate-nAïve subjects investiGating rituximab’s Efficacy) was a randomized, controlled, double-blind trial involving 748 methotrexate-naïve patients assigned to receive rituximab at doses of either 2 × 1000 mg or 2 × 500 mg every 24 weeks in combination with methotrexate or methotrexate alone. The primary end point was radiographic progression measured by total modified Sharp score at week 52. In patients treated with 2 × 1000 mg rituximab and methotrexate, significantly smaller change (0.359) in modified total Sharp score (mTSS) was observed compared with patients on methotrexate alone (1.079; P = <0.001). Furthermore, a significantly higher proportion of patients treated with MabThera and methotrexate had no progression in their joint damage over 1 year (64% vs. 53%; P = .0309). By week 52, 65% of these patients achieved a 50% improvement in symptoms (ACR50) while 47% had achieved a 70% improvement (ACR70), compared with 42% and 25% on methotrexate alone (P < .0001 for both ACR50 and ACR70 comparisons).
Safety Issues
The rapidity and magnitude of peripheral blood B cell depletion following anti-CD20 therapy raise concerns about potential adverse sequelae. The peripheral compartment recovers after many months, but repopulation is predominantly with an immature and naïve subset of B cells. But it must also be remembered that the circulation contains less than 2% of total B cells.31 A common concern regarding all therapies directed at B cells is the potential for toxicity related to modulation of humoral immunity. Unlike other newly introduced biologic therapies for RA, rituximab has the considerable advantage of an oncology safety database based on more than 350,000 non-Hodgkin’s lymphoma patient treatments since 1997.32 The overall safety conclusions are that serious adverse events are infrequent and often associated with well-defined risk factors such as cardiopulmonary disease or a high number of circulating cancer cells. Of note, in the lymphoma population, prolonged peripheral B cell depletion has not been associated with cumulative toxicity or increased occurrence of opportunist infections.33–35 However, it cannot be assumed that the toxicity profile will be identical in distinct disease phenotypes with differing pathogenic processes.
In RA open-label,36 phase II,23,24 and phase III25 studies, although decreases in total serum immunoglobulin levels were observed in patients receiving rituximab, concentrations remained within normal limits. Of note, existing antibody titers against tetanus toxoid appear to be unaffected by a single course of rituximab treatment.37 However, there is some anecdotal evidence that total serum immunoglobulin concentrations fall below the normal range in patients receiving multiple cycles of rituximab treatment over a number of years in open-label studies.11 It is unclear whether this results in an increased risk of infection. In phase II studies, most adverse events were mild to moderate and associated with infusions including headache, nausea, and rigors. In a recent meta-analysis of randomized clinical trial data from three studies reporting adverse events arising after a single cycle of rituximab treatment in a total of 938 RA patients refractory to nonbiologic DMARDs or biologic anti-TNFs,23–25 it was calculated that the incidence of patients experiencing adverse events of all systems was not higher in the rituximab-treated patients than in the placebo groups (relative risk [RR], 1.062; 95% confidence interval [CI], 0.912 to 1.236, P = .438).38
In the DANCER trial, adverse events associated with rituximab were largely associated with the first infusion; these occurred in 39% of patients treated with 500 mg (without steroid) and in 46% receiving 1 g, compared with 17% administered placebo infusions.24 The corresponding incidence with the second infusion decreased to 5%, 8%, and 10%, respectively. Two serious infusion reactions, hypersensitivity and generalized edema, occurred on day 1. Pretreatment with methylprednisolone reduced the incidence and severity of reactions by about one-third (see Figure 64-2). Infectious adverse events (largely upper respiratory tract infections) were reported in 28% of placebo and 35% of rituximab patients. There were six serious infections: two in the placebo group, four in patients receiving 1000 mg rituximab, and none in patients receiving 500 mg rituximab. No opportunistic infections or tuberculosis reactivations were reported.
Although the overall safety record based on trial data has been favorable, with wider clinical use of biologic B cell depletion, rarer serious complications have come to light. A potential association has been reported between the biologic therapies efalizumab, natalizumab, and rituximab and the rare, progressive, and usually fatal condition progressive multifocal leukoencephalopathy (PML), a rare brain disease caused by reactivation of the JC virus.39 PML has been reported in patients receiving rituximab for hematologic conditions and systemic lupus erythematosus (SLE)40 and more recently in RA.39 The cumulative incidence rate of PML in the RA population has been estimated at 1/100,000 RA admissions in an analysis limited to hospitalized patients with SLE and other rheumatic diseases (including 25 patients with RA), a majority of whom had concomitant risk factors including human immunodeficiency virus (HIV), malignancy, or transplantation of bone marrow or another organ.41 The cumulative reporting rate of 2.2 cases of PML per 100,000 RA patients treated with rituximab is more than double the estimated frequency in RA at 2.2 (95% CI, 0.3 to 8.0).39 Although the absolute risk is small, the relative risk is such that it emphasizes the importance of providing the prospective patient being considered for B cell–depleting therapy with thoughtful and balanced information about likely benefits, as well as common to rare complications.
The profound and enduring peripheral B cell depletion that accompanies use of rituximab raises a potential safety concern for those patients in clinical practice who fail to derive adequate symptomatic benefit and may then be exposed to biologic DMARDs of alternative mechanism of action at a time point before repopulation of circulating B cells can take place. Relatively little data regarding this circumstance exist to date, but there is preliminary information in 185 of 2578 RA patients who went on to receive an alternative mechanism of action biologic as documented in a safety follow-up period following participation in trials in which they had previously received rituximab.42 Of the 185 patients 89% remained peripherally B cell depleted at the point of commencing a new mechanism of action biologic. The rate of serious infectious events reported following rituximab but before second biologic exposure was 6.99 per 100 patient-years, comparable with the reported rate of 5.49 per 100 patient-years after exposure to a second biologic agent, the majority of which were TNF inhibitors. No fatal or opportunistic infections were observed, the nature and course of infectious complications being within expectation for RA patients on biologic therapy. In a population of methotrexate-naïve patients entering the IMAGE study,28 safety data were consistent with results from previous rituximab clinical trials and further enhanced the robust safety profile. Rates of serious adverse events and serious infections were similar between the two MabThera groups and the methotrexate-only group.
RA patients, particularly those treated with immunosuppressants, are at an increased risk of infection. For this reason, vaccination is an important aspect of RA clinical management. As to whether B cell–depleting therapy could adversely affect immunization responses by suppressing the antibody response from new vaccination or reducing preformed antibody from prior vaccination has been investigated, and the findings were recently reported.43–45 These studies indicate that vaccine responses to some, but not all, vaccinations may be diminished in rituximab-treated patients, most strikingly in the first 4 to 8 weeks following rituximab administration.43 Although prior vaccination and timing of vaccine administration after rituximab infusion may influence the ability to mount a response, there is no straightforward relationship between peripheral B cell reconstitution and response to immunization. Therefore where vaccination is indicated, it should ideally be given before rituximab treatment and avoided immediately after B cell depletion with a delay of several months. Of course, this may not be practical for vaccines with seasonal availability (such as those to influenza variants), and it must be recognized that responses to vaccination do not necessarily correlate with the risk of infection. In general, the timing of rituximab administration must be determined according to clinical need without requirement to delay until after supplies of a seasonally variable vaccine become available and the patient has been immunized.
In a recent meta-analysis of six studies enrolling 2728 patients, nonmelanotic skin carcinoma occurred more commonly than other carcinomas in the rituximab treatment groups (0.8% in rituximab group vs. none reported in the control group). Moreover, overall incidence of malignancies was higher in the rituximab group (2.1%) compared with the control group (0.6%).46
The safety and efficacy profile of rituximab in the treatment of RA discussed so far is based on reports from randomized placebo-controlled trials of 6 to 12 months’ duration. Open-label extension studies have analyzed safety and efficacy results over multiple courses of rituximab.47 In safety analyses based on 5013 patient-years of rituximab exposure, in a total of 2578 RA patients who received at least 1 course of rituximab, infusion-related reactions were the most common adverse event occurring in a quarter of patients during the first infusion of the initial treatment cycle, although only 1% of infusion reactions in total were considered to be of a serious nature. Importantly and reassuringly, the rates of both adverse events and serious adverse events were stable over infusion cycles, the later reported as 17.85 events per 100 patient-years (95% CI: 16.72, 19.06). Infections and serious infections over time remained stable across five treatment cycles at 4 to 6 events per 100 patient-years. There were no cases of tuberculosis, disseminated fungal infections, or other serious opportunistic infections during the analysis period. Viral reactivation is a potential concern in immunosuppressed patients. The rate of herpes zoster infections in this large series was 0.98 events per 100 patient-years, similar to that reported for other RA populations. The much more serious and rare opportunistic infection, PML occurred in a single patient who also received cancer chemotherapy and radiation. The event occurred about 18 months after the last dose of rituximab and 9 months after receiving chemotherapy and radiation. The causal relationship to rituximab, if any, is thus not entirely clear. There was no increased risk of malignancy by comparison with reference patients with RA and with the general population in the United States. Myocardial infarction was one of the most common serious adverse events reported in the longer-term analyses at a rate of 0.56 per 100 patient-years, but this is consistent with rates reported in epidemiologic studies of patients with RA.
Duration of Benefit
Among RA patients achieving clinical responses to rituximab treatment, the time to clinical relapse is heterogeneous. In some patients, relapse is closely correlated to the reappearance of peripheral blood B cells, but in others, it may be delayed by years.48 Clinical relapse is more closely associated with increases in autoantibody levels, but better biomarkers are necessary to reliably inform optimal management strategies on an individual basis. All B cell populations are depleted following rituximab therapy. Of residual B lineage cells, more than 80% exhibit a memory or plasma cell precursor phenotype.49 B cell repopulation occurs at a mean of 8 months after rituximab therapy and depends on the formation of naïve B cells of an immature phenotype resembling those found in umbilical cord blood. Peripheral B cell depletion is accompanied by substantial increases in blood BLyS concentrations, which tend to fall with B cell repopulation.50 BLyS is a naturally occurring protein required for the development of B lymphocytes into mature plasma cells. Elevated levels of BLyS in RA are believed to contribute to the production of autoantibodies. However, in cases of prolonged clinical responses to rituximab, more gradual reductions in BLyS concentrations have been observed, extending beyond the period of B cell depletion. Thus BLyS may contribute to the survival or regeneration of pathogenic, autoreactive B cells. This hypothesis predicts a potential therapeutic role for BLyS blockade in addition to B cell depletion.
Information concerning the efficacy and safety of repeated cycles of rituximab treatment has emerged from experience in clinical practice and from randomized trials. The recently reported phase III MIRROR trial (Methotrexate Inadequate Responders Randomised study Of Rituximab) was a randomized, double-blind, international study to evaluate the efficacy and safety of three dosing regimens of rituximab in combination with methotrexate in 375 patients with active RA and an inadequate response to methotrexate.51,52 Patients were randomized to three groups with two courses of rituximab treatment at varying doses: Group A: all courses 500 mg rituximab on days 1 and 15; repeat treatment at 24 weeks; group B: first course 500 mg rituximab on days 1 and 15; second course 1000 mg rituximab; repeat treatment at 24 weeks; group C: all courses 1000 mg on days 1 and 15; repeat treatment at 24 weeks. The primary end point was the proportion of patients achieving ACR20 at week 48. Secondary end points included ACR50, ACR70, and EULAR responses. There was a trend toward better efficacy results with the regular 2 × 1000 mg dose compared with the low 2 × 500 mg dose, and this reached statistical significance for EULAR good/moderate response (2 × 1000 mg = 88% vs. 2 × 500 mg = 72%; P < .05). Other end points, although numerically superior at 48 weeks, did not show any statistically significant difference between the three dosing regimens.
Current Role
Rituximab is generally considered as a cost-effective biologic option in RA patients, particularly if seropositive, with inadequate responses to TNF inhibitors. Recent advances in the understanding of the pathogenesis of RA emphasize the critical role of B cells in self-sustaining chronic inflammatory processes. Rituximab is a promising addition to the therapeutic armamentarium for the treatment of RA. In current clinical practice, the major use for rituximab in the treatment of RA is confined to the TNF inhibitor–refractory population. Data from a number of clinical trials (IMAGE, MIRROR, SERENE, REFLEX, and DANCER)24,27,28,51 suggest that seropositive RA patients (RF and/or anticyclic citrullinated peptide [anti-CCP]) show a higher likelihood of response to B cell–depleting therapy compared with seronegative patients, in particular for improving signs and symptoms and inhibition of radiographic changes. However, it is nevertheless the case in both trials and clinical experience that a proportion of seronegative RA patients show good clinical responses, although this proportion is less than in the case of seropositive patients. In pooled analyses of data from the MIRROR and SERENE studies, at week 48, odds ratios for seropositive patients versus seronegative patients of achieving ACR20, ACR50, and ACR70 responses were 2.23 (95% CI, 1.38 to 3.58), 2.72 (95% CI, 1.58 to 4.70), and 3.29 (95% CI, 1.40 to 7.82), respectively.53 These observations generate the hypothesis that other mechanisms may account for lower levels of response in seronegative patients such as antigen presentation, co-stimulation, and cytokine drive, whereas high levels of response to rituximab therapy may be mediated primarily by the suppression of pathogenic antibodies. In contrast to RF, the type I interferon (IFN) signature is associated with a negative response to rituximab therapy in RA patients.54
The optimal and most cost-effective dosing regimen for rituximab remains a matter of debate. The phase III SERENE study showed equal clinical efficacy for the 500 mg × 2 and 1000 mg × 2 rituximab doses,27 but the phase III MIRROR trial51 had differences in some outcomes favoring the higher dosage. Methotrexate-naïve patients (not an approved patient population for rituximab) were studied in the IMAGE study with clinical results that were equivalent, but with a radiographic result that favored the higher dosage.28 Thus a summary of the current randomized controlled trial data on rituximab dosing is that 1000 mg × 2 works well in a clinically meaningful proportion of patients, but not in all. The 500 mg × 2 rituximab dose achieves broadly similar results in the relevant patient populations overall and has the advantages of lower cost and possibly a lower rate of serious adverse events but perhaps with lower probability of high-impact clinical responses and inhibition of structural damage. On the basis of the DANCER study findings, it is recommended that each cycle of 1000 mg × 2 rituximab be given in combination with once-weekly methotrexate, usually at doses of at least 15 mg/week, to optimize efficacy. Further, administration of 100 mg intravenous methylprednisolone is recommended before each rituximab infusion to reduce the frequency and severity of infusion reactions.
Rituximab may also have a role in those patients for whom TNF blockade is relatively contraindicated such as those with connective tissue disease overlap syndromes. At present, there are uncertainties about the implications of long-term peripheral B cell depletion and the timing and need for redosing with rituximab in patients who respond. Current research suggests that restoration of peripheral B cell numbers takes about 8 months after depletion treatment, although retreatment may be necessary earlier. Results have been presented for an open-label study to evaluate the response to repeated courses of rituximab in patients with active RA participating in one of several phase II or III studies and to determine the optimal frequency for repeated treatment.55 In a series of 155 patients with prior exposure to TNF inhibitors, ACR20, ACR50, and ACR70 scores were 65%, 33%, and 12%, respectively, following the first course; they were 72%, 42%, and 21%, respectively, for the second treatment course, relative to the original baseline. In 82 of these patients who received a third course of rituximab, the median interval between first and second courses was similar to that between second and third courses: 30 to 31 weeks.55
Further studies are necessary to identify the optimal regimens for maintenance therapy that will provide efficacy and limit toxicity.56 Development of biomarkers informative of management decisions that would optimize response is a highly desirable goal, but as yet none are in routine use. The magnitude of clinical response appears to be related to the completeness of peripheral B cell depletion. This holds true whether the lower 500 mg × 2 dose schedule or higher 1000 mg × 2 schedule is administered. By means of sensitive measurements permitting detection of low numbers of preplasma B cells in the circulation, a recent study reported that RA patients whose disease did not respond to an initial cycle of rituximab have higher circulating preplasma cell numbers at baseline and incomplete depletion. Furthermore, an additional cycle of rituximab administered before total B cell repopulation enhances B cell depletion and clinical responses.57





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


