27 Cell Survival and Death in Rheumatic Diseases
Cells die in different ways with the morphologic appearance of apoptosis, autophagy, or necrosis.
Different types of cell death dictate different immune responses.


 Supplemental images available on the Expert Consult Premium Edition website.
Supplemental images available on the Expert Consult Premium Edition website.
 Video available on the Expert Consult Premium Edition website.
Video available on the Expert Consult Premium Edition website.
History and Concepts
Apoptosis
Illustrations of cells undergoing apoptosis were made almost as soon as stains were used to examine the appearance of cells in different tissues. Drawings of ovarian follicles undergoing cell death made over 100 years ago demonstrate cell shrinkage and nuclear condensation. Subsequent descriptions of “pyknosis,” chromatin margination, and other terms used to convey the appearance of subcellular particles during cell death included many features now recognized as apoptosis. The history of this subject is reviewed elsewhere.1
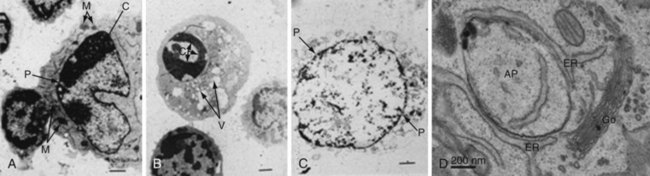
Our modern understanding of apoptosis began with electron microscopic descriptions of morphologic changes characterized by shrinkage of hepatocytes (i.e., shrinkage necrosis) after ischemic or toxic injury to the liver. The name apoptosis was coined by Kerr, Wyllie, and Currie in 1972 to describe the form of death that was “consistent with an active, inherently controlled phenomenon” characterized by cell shrinkage, nuclear condensation, and cell blebbing (Figure 27-1).2 This term also conveyed the concept of cell death that was similar to leaves falling from a tree (apo means “from,” and ptosis, “a fall,” in Greek), implying a regulated “mechanism of cell deletion, which is complementary to mitosis.”2
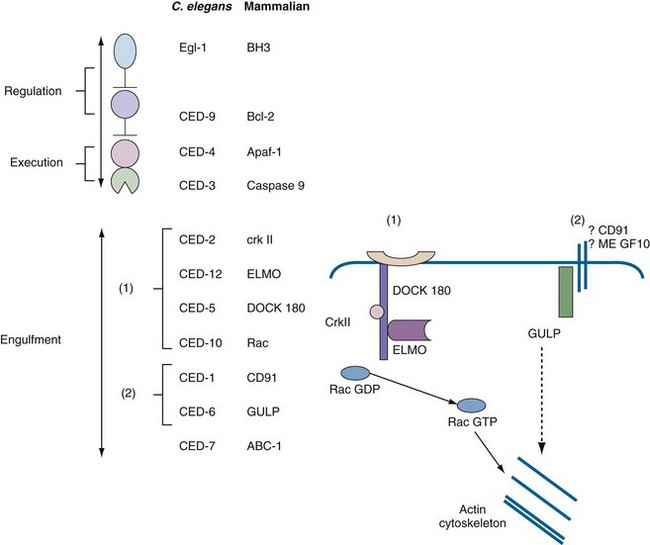
Further developments in apoptosis paralleled advances in molecular biology, genetics, and biochemistry. The detection of a nucleosomal ladder3 was of considerable importance, because it defined a biochemical event (i.e., nucleosomal cleavage) and provided a simple electrophoretic test for detection of apoptotic cell death that remains a standard in the field (see Laboratory Detection of Apoptosis later in this chapter). Another landmark was the discovery in the 1980s that the death of cells during development of the nematode Caenorhabditis elegans was under strict genetic control. Remarkably, the death of these cells could be perturbed by mutations of a small number of genes called ced (for cell death abnormal) genes.4 Horvitz and colleagues determined that two ced genes, ced3 and ced4, encoded death effectors, whereas ced9 was an antiapoptotic gene.4 Most of the remaining ced genes were responsible for engulfment and removal of “corpses.” This simple model in which CED-3 is the main death protease that is activated by CED-4 and inhibited by CED-9 has served as a paradigm for defining apoptotic pathways in mammalian cells (Figure 27-2). In 2002, Robert Horvitz was awarded the Nobel Prize for discoveries concerning the genetic regulation of programmed cell death.
Mammalian cells are much more complex and, as will be discussed in detail, have multiple defined pathways that follow the basic C. elegans model. The molecules within these pathways, the downstream effectors of apoptosis, the caspases (cysteine aspartate proteases), and the proteins implicated in the clearance of apoptotic cells are discussed in detail here. Regulation of cell death is of seminal importance in a number of diseases, including cancers, autoimmune diseases, and degenerative disorders.5,6 The relevance of apoptosis to rheumatic disorders is summarized at the end of the chapter.
Autophagy
Autophagy, which means “to eat oneself,” is essentially a protective process that occurs after growth factor withdrawal or nutrient depletion that may or may not result in cell death. The process is highly conserved from yeast through humans. Under circumstances of nutrient depletion, cells switch to a catabolic program in which cellular constituents are degraded for energy production as a survival mechanism. In these cells, a double membrane vesicle (autophagosome) forms around organelles or protein aggregates and encapsulates them (see Figure 27-1D). A characteristic feature of this process is the lipidation of LC3 (microtubule-associated protein 1 light chain 3) to form LC3-II, which can be identified as coarse dots on the autophagosomes seen by microscopy (see Figure 27-6I, later), or by changes in molecular mass on Western blot analysis. The autophagosomes then fuse with the lysosomes, leading to degradation of their contents (Figure 27-3).
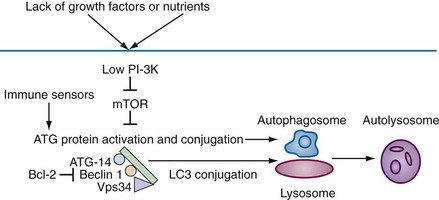
The biochemical pathways by which autophagy is initiated and executed are complex and are schematically outlined in Figure 27-3. In brief, lack of nutrients leads to a reduction in phosphoinositide (PI)-3 kinase activity, resulting in loss of suppression of mTOR (mammalian target of rapamycin) and activation of autophagic (ATG) proteins. The ATG gene family comprises at least 20 members and includes beclin1 (ATG6), a protein that is critically involved in the regulation of autophagic cell death.7 Beclin1 is monoallelically deleted in a variety of human cancers, and may function in control of cell growth and in tumor suppression.8 Autophagic death shares a number of morphologic features with necrotic cell death9 and may be seen in neuronal cell death associated with polyglutamine repeats. Many reports indicate that autophagy is important in immune function. Examples include intracellular host response to pathogens, survival of lymphocytes after growth factor withdrawal, Toll-like receptor (TLR) stimulation in plasmacytoid dendritic cells (pDCs), and major histocompatibility complex (MHC) class II presentation of antigen.10,11 The strongest link between autophagy and autoimmunity or autoinflammatory disease is the association between a genetic variant of ATG16L1 and another protein that regulates autophagy, the immunity-related guanosine triphosphatase gene (IRGM), and Crohn’s disease. The precise mechanisms that account for this association remain to be determined.11
Necrosis
Necrosis (from the Greek nekros, meaning “corpse”) is distinguished from apoptosis predominantly by morphologic appearance.12 Necrotic cells are swollen, and electron microscopy reveals disorderly fragmentation of chromatin and severe damage to the mitochondria (see Figure 27-1). The cellular membrane loses integrity and becomes permeable to vital dyes such as trypan blue and propidium iodide (see Figure 27-6, later). The distinction between apoptosis and necrosis remains important from a number of perspectives. In contrast to the genetic and biochemical programs that regulate apoptosis, necrotic cells usually result from death “by accident,” that is, from thermal or drug injury, infection, or infarction of an organ. Because of uncontrolled release of intracellular contents, necrotic cells induce a proinflammatory immune response, whereas apoptotic cells usually elicit an anti-inflammatory response.
The same inducers (e.g., ischemia, hydrogen peroxide) may produce apoptosis or necrosis, depending on the severity of the injury and the rapidity of cell death. The fate of the cell is determined in part by cellular energy reserves, especially adenosine triphosphate (ATP).13 ATP is generated by oxidative phosphorylation in mitochondria, as well as by glycolysis in the cytosol. Some inducers initially may cause apoptosis followed by necrosis (postapoptotic necrosis). This is likely to occur when removal of apoptotic cells is delayed and is thought to be important in stimulating an inflammatory response to self-antigens. A unique type of necrosis in neutrophils, called NETosis, is implicated in SLE13a (see Defective Uptake and Processing of Apoptotic Cells).
Pyroptosis
Pyroptosis (from the Greek pyro, meaning “fire”) is distinguished from other forms of cell death first and foremost by the activation of caspase 1 (interleukin [IL]-1β–converting enzyme) and secretion of the inflammatory cytokine, IL-1β. Pyroptosis is most strongly associated with infection by intracellular bacteria such as Salmonella, Yersinia, and Legionella, although it may also be seen following tissue infarction.14 Cells undergoing pyroptosis demonstrate nuclear condensation associated with DNA damage, cell swelling, and ultimately cell lysis associated with release of IL-1β.14 The mechanisms responsible for this process involve intracellular sensors of bacterial products and formation of the inflammasome. These topics are briefly described in the following sections and elsewhere in the text (see Chapter 18).
Biochemistry of Apoptosis
A schematic diagram of the cell death program is shown in Figure 27-4. A brief outline of each major functional component within the program, from signals for death to removal of apoptotic cells, is provided here, but space limitations preclude a detailed analysis of the layers of regulation at each step of the pathway. For example, post-translational protein modifications such as phosphorylation, nitrosylation, and oxidation present additional complexities that are under intense study. A series of reviews in Apoptosis and Its Relevance to Autoimmunity15 offer more detailed information on the biochemistry of apoptotic pathways and their relationship to immune function.
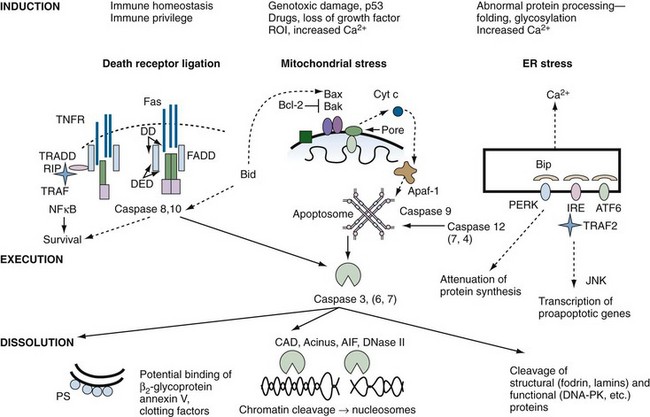
The specialized proteins involved in apoptosis and its regulation contain a number of modules or domains that are predominantly involved in promoting protein-protein interactions (Table 27-1). These domains may be found in receptors, adapters, effectors, or inhibitors. Furthermore, as will be discussed later, these domains occur in proteins involved in apoptosis, as well as inflammation (Supplemental Figure 27-1 on www.expertconsult.com). It has been suggested that death domain (DD), death effector domain (DED), caspase recruitment domain (CARD), and pyrin domain all evolved from the prototypic DD-fold, corresponding to an antiparallel six-helix bundle.16 In general, like domains bind so as to facilitate homotypic interactions, leading to oligomerization of the same protein or binding to different proteins in a signaling pathway. These changes usually produce conformational alterations, which lead to further protein recruitment. Other domains such as the nucleotide-binding site (NBS) specify nucleotide binding.
Table 27-1 Modular Components of Proteins Involved in Apoptosis and Inflammation
| Module* | Component of | Function |
|---|---|---|
| BH (1-4) | Bcl-2 family | P-P-I |
| BIR | IAP family | P-P-I |
| CARD | Caspases, adapters, NODs | P-P-I |
| DD | Death receptors, adapters, kinases | P-P-I |
| DED | Adapters, caspases | P-P-I |
| LRR | Pyrin family, NODs, TLR | P-P-I |
| NBS/NOD | Pyrin family, NODs | Nucleotide-binding, oligomerization |
| Pyrin | Pyrin family | P-P-I |
| RING finger | IAP family | E3-ubiquitin ligase |
BH, Bcl-2 homology; BIR, baculovirus AIP repeat; CARD, caspase recruitment domain; DD, death domain; DED, death effector domain; IAP, inhibitor of apoptosis; LRRs, leucine-rich repeats; NBS, nucleotide-binding site; NOD, nucleotide oligomerization domain; P-P-I, protein-protein interaction; TLR, Toll-like receptor.
* See Mariathasan and Monack180 for further discussion.
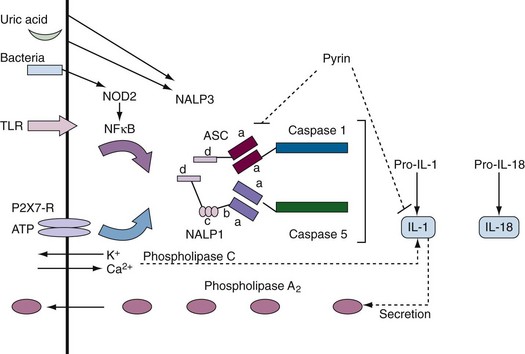
Supplemental Figure 27-1 Components of inflammasomes. Inflammasomes contain pro-interleukin (IL)-1 but different sensors and regulators. In most cases, two stimuli (e.g., activation of a Toll-like receptor [TLR] or nucleotide-binding oligomerization domain [NOD] sensor plus potassium flux) are required to activate the inflammasome.51 The prototypic inflammasome containing caspase 1 or 5 (caspase 11 or 12 in mice) and the adapter protein, ASC (Pycard), is shown. A NALP3-containing inflammasome is of rheumatologic interest because it is activated by uric acid and certain nucleic acids. NOD2 is mutated in Crohn’s disease and Blau syndrome. Pyrin is mutated in familial Mediterranean fever (FMF). The domain structure of these proteins (see Table 27-1) is as follows: a = CARD domain; b = nucleotide-binding site; c = leucine-rich repeat; d = pyrin domain.
Death Ligands, Receptors, and Signals
Death of a cell may result from intracellular stress activating an intrinsic death program, or it may be forced on the cell by the interaction of a death ligand with a death receptor (see Figure 27-4). Death receptors (DRs) belong to the tumor necrosis factor (TNF) receptor (TNFR) superfamily of proteins, which comprises approximately 25 members.17,18 This family of receptors is responsible for diverse biologic responses such as inflammation, proliferation, antiviral activity, and cell death. Receptors in the TNF family include at least six receptors capable of transmitting apoptosis (see later), as well as receptors such as CD40, CD30, BlyS/BAFF/TALL, and TACI,17 which trigger survival and/or proliferation in part through activation of nuclear factor κB (NFκB). Although most receptors of the TNFR family exert their effects primarily within the immune system, some members (e.g., p75NGFR, TNFR I and II) appear to serve important functions in the nervous system and in other organs. Some TNFR-like proteins, such as PV-T2, PV-A53R, and CAR1, are encoded by viruses and may contribute to their virulence.17
Death Receptors
The DRs identified, including Fas, TNFR I, DR-3 (TRAMP/wsl/APO-3), DR4 (TRAIL, receptor for the Apo2 ligand), DR5, and DR6, share homology in their intracellular domains over a 70 amino acid region called the death domain (DD).19 Three decoy receptors have been identified—two (DcR1 and DcR2) that bind and inhibit their ligand, TRAIL, and one (DcR3) that binds Fas ligand. These decoy receptors presumably modulate the cytotoxic function of the ligands, but their biologic contexts remain to be fully defined. Use of alternative splice forms and shedding of the receptors and ligands also downmodulate their function.
TNFR family members are characterized by two to six cysteine-rich domains (CRDs) in their extracellular regions.17 The co-crystal structure of TNFR I and lymphotoxin-α indicates that CRDs project from the cell surface in a linear array, making distinct contact with ligands at subunit interfaces. The first CRD may also be responsible for preassembly of the receptor as trimers that undergo further conformational alteration upon ligand engagement.
The three-dimensional structure of the DD has been solved by nuclear magnetic resonance spectroscopy and has been shown to consist of six amphipathic α-helices that create a unique fold.20 Functionally, the DD appears to be a novel protein-protein association motif that facilitates homotypic interactions. For example, the DD of Fas and TNFR I self-associate, thereby recruiting adapter proteins that also contain DD and that directly or indirectly mediate receptor signal transduction (see Figure 27-4).
Death Receptor Signal Transduction
This section will focus predominantly on signaling from Fas and TNFR, because these are the best-characterized members of the death receptor subfamily, and it is likely that other death receptors signal through similar pathways. As illustrated in Figure 27-4, Fas and TNFR I share a common death pathway. Binding of Fas ligand to Fas causes conformational changes in the receptor cluster, leading to recruitment of intracellular adapter molecules. Initially, aggregation of Fas induces uptake of the adapter protein, FADD, to the DD of Fas. FADD has two structural domains: a C-terminal DD, which mediates Fas binding, and an N-terminal death effector domain (DED). The FADD DED allows recruitment of procaspase 8 and procaspase 10 through DED-DED interactions. Procaspases 8 and 10 have a bipartite structure comprising a DED and an enzymatic caspase domain, the latter linking Fas aggregation with the execution phase of apoptosis. Apposition of procaspases 8 and 10 to the activated Fas complex leads to autocatalytic cleavage and conversion of the proenzymes to activated proteases, which are released and are able to initiate a proteolytic cascade, leading to programmed cell death. In some cell lines, caspase 8 cleavage also results in cleavage of the proapoptotic molecule Bid, which activates the mitochondrial amplification cascade (type II pathway21; Supplemental Figure 27-2 on www.expertconsult.com; see also Figure 27-4).
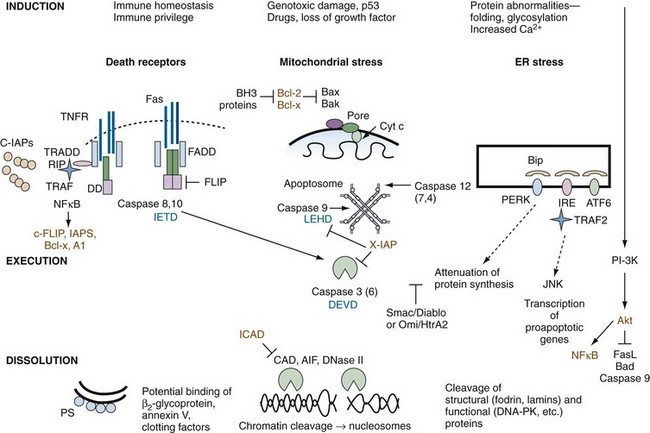
Supplemental Figure 27-2 Inhibitors of apoptosis. Inhibitors of death pathways are shown in brown. Note that a number of tumor necrosis factor (TNF) family members (e.g., CD40L, BAFF/ BLyS/TALL, receptor activator of NFκB [RANK] ligand) activate the nuclear factor κB (NFκB) pathway, resulting in increased transcription of antiapoptotic proteins, inhibitors of apoptosis (IAPs), Bcl-2 family members, and A1.181 In some cases, inhibitors of apoptosis are blocked or destroyed during apoptosis. For example, the protein Smac/Diablo is released from mitochondria and eliminates the antiapoptotic effect of the IAP family. Growth factors impart survival signals, in part through the phosphoinositide (PI)-3 kinase → Akt pathway (right side of diagram). Activation of the kinase, Akt, directly inactivates certain proapoptotic proteins such as Bad and caspase 9, and promotes survival by inhibiting the forkhead family (FKRL) of transcription factors and enhancing the expression of the antiapoptotic protein, NFκB.182 Inhibitor of caspase-activated DNase (ICAD) is a specific inhibitor of the DNAse, CAD, which is degraded by caspases. Tetrapeptide inhibitors of specific caspases are shown in blue.
Defective function of caspase 8 leads to a form of cell death called NECROPTOSIS that combines features of apoptosis and necrosis resulting in activation of NFκB and production of inflammatory cytokines such as TNF.21a
Death Ligands
FasL (CD178) is a 40-kD type II transmembrane protein that shares 15% to 35% amino acid identity with the TNF superfamily of ligands. FasL is expressed constitutively in the anterior chamber of the eye and in the testis but is induced when CD8, T helper-1 (Th1) CD4+ T cell, and some natural killer (NK) cell populations become activated.22 In lymphocytes, expression of ligand is tightly regulated and activity on the cell surface short-lived, because metalloproteases cleave the extracellular portion of the ligand into soluble, functional molecules. The zinc metalloprotease, TNF-converting enzyme (TACE), is a membrane-anchored member of the disintegrin family of proteases that cleave active TNF from the cell surface.23,24
Function in Immune Regulation
Although Fas and FasL interactions in the thymus are not thought to play a major role in negative selection, this pathway is involved in the maintenance of immune privilege in the eye and the testis, in the pathogenesis of graft-versus-host disease, and in immune evasion by tumor.22 The major physiologic function of Fas and FasL in the immune system is the preservation of peripheral tolerance. This is achieved by the phenomenon of activation-induced cell death (AICD), whereby CD8 T cells, Th1 CD4+ T cells, and possibly NK cells induce apoptosis of activated T cells, B cells, and macrophages. Deletion of activated immune cells removes the source of proinflammatory molecules, prevents the continued presentation of self-peptides by primed (high levels of co-stimulatory molecules) antigen-presenting cells, and eliminates B cells that have mutated to self-specificity within the germinal centers.25 More recently, it has been shown that Fas-FasL interactions promote a variety of additional functions, including early T cell proliferation, tumor survival, and cell migration. These topics are discussed in greater detail elsewhere,26,27 and the consequences of Fas deficiency are described later in this chapter.
It should be noted that, whereas TRAIL signals apoptosis through DR4 and DR5 predominantly in tumor cells, evidence suggests that TRAIL also plays a role in negative selection of thymocytes.28 Similarly, DR3 (TWEAK, receptor for the Apo3 ligand) has been implicated in negative selection.29 Finally, DR6 plays a role in immunologic homeostasis, as evidenced by enhanced T and B cell proliferation in DR6-deficient mice.30
Intrinsic Cell Death Pathways: Initiation and Execution of Apoptosis
Cells need constant sources of nutrition and depend on a variety of signals for active maintenance of survival. Loss of signals from neighboring cells31 or withdrawal of growth factors or cytokines results in initiation of a cell death program. Damage or stress to intracellular organelles may be induced from outside or within the cell. Here, injury or stress to DNA, mitochondria, and the endoplasmic reticulum is discussed.
Genotoxic Injury
Mutations occur frequently in mammalian DNA and usually are promptly repaired. However, if repair fails or DNA is severely damaged by radiation or drugs, the transcription factor, p53 (“guardian of the genome”), is upregulated and phosphorylated by DNA damage sensors such as ATR and ATM. Activated p53 induces cell cycle arrest through induction of the cyclin-dependent kinase inhibitor, p21. If DNA damage is repaired, cell cycle arrest is abrogated, whereas if the injury cannot be repaired, the cell undergoes apoptosis. The critical importance of p53 as a tumor suppressor is illustrated by the high frequency of p53 mutations in cancers.32 p53 induces apoptosis, in part through transcription of death effectors such as Bax that cause mitochondrial stress. In addition, activation of the transcription factor Foxo-1 upregulates the expression of Bim, FasL, and TRAIL.33
Mitochondrial Stress
Mitochondria are cytoplasmic organelles that contain their own 16-kb genome encased by inner and outer membranes, with a number of proteins, including cytochrome-c, situated between these membranes (see Figure 27-4). Mitochondria help to maintain redox potential and serve as the energy powerhouse of the cell through the generation of ATP by oxidative phosphorylation. These biochemical pathways create an electrochemical gradient (Δψ) that is positive and acidic on the outside and alkaline on the inner side of the mitochondrial membrane. Spanning the inner membrane is the adenine nuclear translocator (ANT), which mediates ATP transport (with VDAC, see later) to the cytosol. On the outer mitochondrial membrane (OMM) is situated the voltage-dependent anion channel (VDAC), which is permeable to solutes of approximately 5000 kD.34
Once MMP is initiated, cytochrome-c is released from the intermitochondrial space into the cytosol (see Figure 27-4). In the cytosol, cytochrome-c and the cofactors Apaf-1 and ATP or dATP assemble with caspase 9 to form a molecular aggregate called the apoptosome, which promotes the cleavage of procaspase 9 into its active form.35 Caspase 9 acts on effector caspases such as caspase 3, resulting in the caspase cascade that leads to cleavage and inactivation of a wide variety of substrates within the cell (see Figure 27-4). A caspase-independent apoptosis-inducing factor (AIF) is also released from the mitochondria and induces nuclear changes and cell death by less well defined pathways.36
Endoplasmic Reticulum (ER) Stress
The main functions of the ER are to regulate intracellular calcium flux and to promote proper folding of nascent proteins. In the contiguous conduit, the Golgi apparatus, post-translation modifications such as glycosylation and isoprenylation are executed. Elaborate mechanisms are in place to ensure that errors in protein folding do not occur, but if they do, an unfolded protein response (UPR) is initiated. The ER/Golgi initiates apoptosis if calcium flux is excessive, if unfolded proteins persist, or if post-translational protein modification is abnormal.37 Apoptosis is the result of three central players: inositol-requiring protein-1 (IRE1), activating transcription factor-6 (ATF6), and protein kinase RNA-like ER kinase (PERK) (see Figure 27-4). Unfolded proteins may activate these proteins directly, or by binding to the ER chaperone immunoglobulin-binding protein, Bip. Once activated, the three proteins cause selective RNA cleavage, termination of protein synthesis, and induction of proapoptotic responses (see Figure 27-4). In contrast to the death receptor or mitochondrial pathways, apoptosis is executed through caspase 12 in mice and possibly caspase 4 in mammals.38 However, many of the same molecules that regulate apoptosis in the mitochondria, the Bcl-2 family in particular, also influence ER-mediated apoptosis, possibly through control of intraluminal calcium. It is important to point out that induction of the UPR, in certain circumstances, leads to NFκB activation and inflammation, rather than cell death.39
Antiapoptotic Proteins: FLIP, Bcl-2, IAPs, and Akt
(See Supplemental Figure 27-2 on www.expertconsult.com.)
Inhibition of Death Receptors
In most resting cell types that express Fas on their cell surface (e.g., lymphocytes), the receptor is nonfunctional. Resistance to death is explained both by low levels of expression of the receptor and by active inhibition by a protein called FLIP. FLIP resembles the structure of caspase 8 and competes with caspase 8 for recruitment to FADD. This prevents FADD from initiating apoptosis. When lymphocytes become activated, FLIP is usually degraded, allowing Fas signal transduction to occur unimpeded (see Supplemental Figure 27-2 on www.expertconsult.com). Similarly, a protein called SODD (silencer of death domain) attenuates TNFR I signal transduction.
Bcl-2 Family of Cell Death Regulators
The Bcl family comprises more than 18 members.40 Bcl-2 is the prototype antiapoptotic protein that was first discovered to be overexpressed in certain B cell lymphomas (Bcl). Of particular significance, Bcl-2 overexpression did not enhance cell proliferation (most cells were in the G0/G1 phase of the cell cycle) but rendered the cells more resistant to death. The antiapoptotic members—Bcl-2, Bcl-XL, Bcl-w, Mcl-1, and A1—contain three or four of the characteristic Bcl-2 homology (BH) domain motifs and most possess the N-terminal BH4 domain and the hydrophobic C-terminal membrane anchor, accounting for their attachment to mitochondrial, ER, and nuclear membranes (see Supplemental Figure 27-2 on www.expertconsult.com). Virus-encoded proteins—BHRF1, LMW5-HL, ORF16, KS-Bcl-2, and E1B-19K—and Bcl-2 have similar antiapoptotic functional properties. The proapoptotic members of this family can be subdivided into two groups: the Bax/Bak-like proteins (Bax, Bak, Bok, and BCl-Xs), which contain two to three BH3 domains, and the BH3-only subset (Bad, Bik, Bid, Hrk, Bim, Noxa, Puma, and Bmf), which contain only the single domain. Bcls are regulated at transcriptional and post-transcriptional levels by a multitude of stimuli.
How do Bcls regulate apoptosis? One level of regulation is conferred by binding interactions (homodimerization or heterodimerization) between members via their BH1, BH2, and BH3 domains.41 Although outcomes vary for each specific pair, homodimerization of Bcl-2 or Bax potentiates their antiapoptotic or proapoptotic functions, respectively, whereas heterodimers may potentiate or abrogate function of one member of the pair. Bax and Bak have been shown to be pivotal downstream effectors of intrinsic apoptotic pathways. A possible model is that Bcl-2 (or homologues) usually heterodimerizes with Bax and Bak, preventing apoptosis. Increased expression of a BH3 proapoptotic protein causes binding to Bcl-2, thereby releasing Bax and Bak to induce apoptosis. It has also been suggested that BH3 proteins may bind and directly activate Bax/Bak.
Bcl regulation of cell death is closely connected to mitochondrial function. The physical association of Bcl-2 family proteins with the outer mitochondrial membrane, as well as the close structural similarity between BH1 and BH2 domains and bacterial pore-forming proteins such as colicin,42 allows them to regulate ion fluxes or the transfer of small molecules from the membrane. In vitro models suggest that Bax and Bak promote opening of the VDAC, allowing the release of cytochrome-c into the cytosol, whereas Bcl-2 binds directly to VDAC and closes it.34
Intracellular Inhibitors of Apoptosis (IAPs)
Intracellular inhibitors of apoptosis (IAPs) are a separate family of antiapoptotic proteins that are highly conserved through evolution. The neuronal apoptosis inhibitory protein (NAIP) was discovered through association of NAIP mutations in patients with the severe form of spinal muscular atrophy. Seven additional members of the family (c-IAP-1, -2, X-IAP, survivin, ILP2, ML-IAP, and Bruce) that share a baculovirus IAP repeat (BIR) domain have subsequently been identified; most contain a RING domain that functions as an E3 ligase. Ubiquitylation may target interacting proteins for proteasomal degradation, or they may be activated. IAPs such as X-IAP directly inhibit effector caspases, especially caspase 9 (see Supplemental Figure 27-2 on www.expertconsult.com), whereas cIAPs modulate cell survival through ubiquitylation of substrates such as RIP and proteins in the NFκB pathway (see Supplemental Figure 27-2 on www.expertconsult.com).43 IAPs block apoptosis induced by a variety of stimuli, including Fas, TNF, ultraviolet irradiation, and serum withdrawal, and survivin is overexpressed in certain cancers and in the rheumatoid arthritis (RA) synovium.44 In some cells, the antiapoptotic effect of IAPs is eliminated by release of the protein Smac/Diablo or Omni/HtrA2 from the mitochondria (see Supplemental Figure 27-2 on www.expertconsult.com).
Akt
Akt is a cytosolic protein kinase (protein kinase B) that serves a special role in prevention of apoptosis, because it links cell activation through PI-3 kinase with multiple transcription factors. Phosphorylation of Akt is antagonized by the phosphatase, PTEN. When phosphorylated, Akt promotes cell survival by altering the function of intrinsic (mitochondrial) and extrinsic (death receptor) pathways of apoptosis. Specifically, this includes inactivation of proapoptotic molecules such as caspase 9 and Bad, and activation of survival pathways that include NFκB and forkhead transcription factors that inhibit FasL (see Supplemental Figure 27-2 on www.expertconsult.com).45 Antiapoptotic molecules may be cell type or context specific.
Caspases
Caspases are cysteine-containing proteases that have an unusual substrate specificity for peptidyl sequences with a P1 aspartate residue.46,47 These proteases are 30 to 50 kD in size and comprise an amino-terminal prodomain with a large subunit domain and a small subunit domain (see Figure 27-4). Active site cysteine residue is contained within the conserved pentapeptide, QACxG, on the large subunit of the enzyme, whereas most of the substrate specificity is determined by the small subunit. The upstream caspases 8, 9, 10, 2, and 4 have large prodomains that interact with regulatory proteins such as FADD for caspases 8 and 10 and Apaf-1 for caspase 9 (see Figure 27-4). Clustering of these complexes allows autocatalytic cleavage of large and small subdomains to form the active tetramer. Effector caspases such as 3, 6, and 7 have small prodomains and are thought to be cleaved into their active forms by the upstream caspases.
Members of the caspase family can be divided into three functional subgroups on the basis of their substrate specificities.48 Group I members (caspases 1, 4, and 5) are potently inhibited by the serpin CrmA; group II members (caspases 2, 3, and 7) are specific for DExD; and group III members (caspases 6, 8, 9, and 10) are specific for I/V/LExD—a sequence that is also contained at the junctions of the caspase subunits themselves. Significantly, granzyme B produced by cytotoxic T cells has a substrate specificity similar to that of group III caspases and is capable of inducing apoptosis through this pathway. Identification of the substrate specificity of caspases has led to a number of practical applications, including the ability to quantify activity using fluorogenic tetrapeptide substrates and blockade of proteolytic activity with noncleavable cell-permeable tetrapeptide analogues (see Supplemental Figure 27-2 on www.expertconsult.com).
Effector caspases are necessary for the execution of apoptosis. They cleave specific substrates such as the structural proteins fodrin, gelsolin, and lamins, which are key intracellular enzymes involved in DNA repair (e.g., poly ADP ribose polymerase, DNA-PK) (see Figures 27-4 and 27-6 [later]). These changes facilitate inactivation of synthetic functions of the cell, dissolution of the nuclear membrane, and packaging of cellular proteins into apoptotic blebs on the cell surface. Caspases also cleave regulatory proteins such as Bcl family members and the inhibitor of caspase-activated DNase (ICAD). Cleavage of ICAD leads to the release of active CAD, which enters the nucleus and cleaves nucleosomes at the linker region, yielding the characteristic “DNA ladder” (see Figures 27-4 and 27-6 [later]).49,50
As was already mentioned in the section on pyroptosis, not all caspases are involved in the execution of apoptosis. Human caspases 1, 4, 5, and 12 and mouse caspases 1, 4, 11, and 12 are most likely involved in inflammation. Caspase 1 and caspase 5 interact to form a multiprotein complex that has been called the inflammasome51 (analogous to binding of the apoptosome caspases 1 and 5 to the adapter proteins, ASC [PYCARD] and NALP1 [DECAP], respectively, by their CARD domains) (see Supplemental Figure 27-1 on www.expertconsult.com). ASC and NALP1 are multidomain proteins that contain many of the protein interaction domains listed in Table 27-1. The N-terminus of the protein, pyrin, which is mutated in familial Mediterranean fever (FMF), binds to ASC. The C-terminus of pyrin, which is mutated in FMF, binds directly to IL-1β, suggesting that pyrin normally directly exerts an inhibitory effect on IL-1β.52
Caspases are tightly regulated by their own prodomains and by Bcl and IAP family members (see Supplemental Figure 27-2 on www.expertconsult.com). In addition, viral proteins such as serpin CrmA, produced by cowpox, and p35, produced by baculovirus, are potent inhibitors of caspases.
Finding, Removing, and Responding to Dead and Dying Cells
Finding the Dying Cell
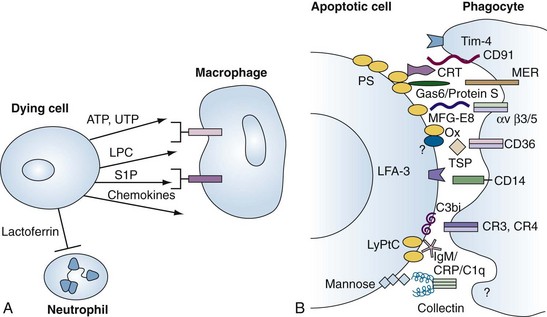
During apoptosis, the enzyme, calcium-independent phospholipase A2, is activated by caspase cleavage, leading to the generation of lysophosphatidylcholine (LPC) (see Figure 27-5 for complete apoptosis schema). LPC acts both as a soluble chemoattractant for macrophages and as an epitope on the cell membrane for natural immunoglobulin (Ig)M antibodies.53 Additional “find-me” signals include sphingosine-1-phosphate (S1P) and the nucleotides ATP and UTP, which are released by dying cells, especially as the plasma membrane becomes damaged (necrosis).54,55 Intravital microscopy coupled with biochemical studies revealed that cell necrosis led to release of ATP, activation of Nrlp3 inflammasomes, and release of IL-1β.56 Subsequently, a chemokine gradient attracted neutrophils to the site of cell damage and formylated peptides to the necrotic cell.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


