89 Antineutrophil Cytoplasm Antibody–Associated Vasculitis
Classification of This Group of Vasculitides
The primary systemic vasculitides involve small and medium-sized vessels and are associated with autoantibodies that target neutrophil cytoplasm antigens (antineutrophil cytoplasm antibodies [ANCAs]). Thus they are frequently referred to as ANCA-associated vasculitides (AAVs), although the terminology ANCA disease has also been proposed.1 The presence of ANCA suggests that AAVs are autoimmune diseases. Individual disease descriptions include granulomatosis with polyangiitis (GPA), formerly known as Wegener’s granulomatosis; microscopic polyangiitis (MPA); allergic granulomatosis with polyangiitis (AGPA), formerly known as Churg-Strauss syndrome; and renal-limited pauci-immune necrotizing and crescentic glomerulonephritis (RLV). The name changes have been recommended by the Boards of the ACR, the American Society of Nephrology, and EULAR, which wished a shift from eponyms to disease-descriptive or cause-based nomenclature.
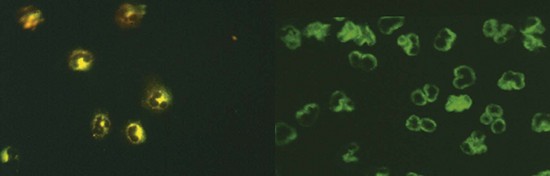
The major autoantigens to which ANCAs are directed within neutrophils and monocytes include two enzyme proteins identified in the 1980s as autoantigens, namely, proteinase-3 (PR3), myeloperoxidase (MPO) (Figure 89-1), and, more recently, lysosome-associated membrane protein 2 (LAMP2).2–4 Confirmation of the presence of antibodies directed against LAMP2 in AAV is awaited, so this antibody subclass is not routinely tested for in current clinical practice.
The initial descriptions of GPA, MPA, and AGPA occurred in the 1930s, 1940s, and 1950s, respectively. However, the term polyarteritis nodosa (PAN) was often used during this period in a generic manner to cover these conditions, particularly MPA, despite the fact that Kussmaul and Meyer had clearly described PAN in 1866 as a systemic condition in which inflammation of medium-sized muscular arteries occurs, leading to aneurysm formation and ischemic tissue or organ infarction.5 The AAVs, in contrast, are generally considered disorders affecting both small and medium-sized arteries. In 1990, the ACR published classification criteria for seven types of vasculitis that included GPA and AGPA, but not MPA, thereby helping to begin to unravel the complexities of vasculitis classification. Four criteria were selected for GPA:
• Abnormal urinary sediment (red cell casts or greater than five red blood cells per high-power field)
• Abnormal findings on chest radiograph (nodules, cavities, or fixed infiltrates)
• Oral ulcers or nasal discharge
Six criteria were selected for AGPA:
2. Eosinophilia greater than 10% on differential blood cell count
3. Mononeuropathy (including multiplex) or polyneuropathy
4. Nonfixed pulmonary infiltrates on chest radiograph
5. Paranasal sinus abnormality
6. Biopsy containing a blood vessel with extravascular eosinophils7
The next major step forward was development of definitions for forms of vasculitis by the Chapel Hill Consensus Conference (CHCC) in 1994, which included GPA, MPA, and AGPA in the 10 types of vasculitis considered.8 These are shown in totality in Table 89-1. Neither the ACR classification criteria nor the CHCC definitions included ANCA as a criterion. Lack of classification criteria for MPA prompted development of an algorithm by international consensus that has been validated in two separate populations, covering GPA, MPA, and AGPA, as well as PAN.9,10
Table 89-1 Classification of Vasculitis as Adopted by the Chapel Hill Consensus Conference on the Nomenclature of Systemic Vasculitis*
| Histopathology | Comments | |
|---|---|---|
| Large Vessel Vasculitis | ||
| Giant cell (temporal) arteritis | Granulomatous arteritis of the aorta and its major branches, with a predilection for the extracranial branches of the carotid artery | |
| Takayasu’s arteritis | Granulomatous inflammation of the aorta and its major branches | |
| Medium-Sized Vessel Vasculitis | ||
| Polyarteritis nodosa† (classic polyarteritis nodosa) | Necrotizing inflammation of medium-sized or small arteries without glomerulonephritis or vasculitis in arterioles, capillaries, or venules | |
| Kawasaki disease | Arteritis involving large, medium-sized, and small arteries, associated with mucocutaneous lymph node syndrome | |
| Small Vessel Vasculitis | ||
| Granulomatosis with polyangiitis‡ | Granulomatous inflammation involving the respiratory tract and necrotizing vasculitis affecting small to medium-sized vessels (capillaries, venules, arterioles, arteries) | |
| Allergic granulomatosis with polyangiitis‡ | Eosinophil-rich and granulomatous inflammation involving the respiratory tract and necrotizing vasculitis affecting small to medium-sized vessels, associated with asthma and blood eosinophilia | |
| Microscopic polyangiitis‡ | Necrotizing vasculitis with few or no immune deposits affecting small vessels (capillaries, venules, arterioles) | |
| Henoch-Schönlein purpura | Vasculitis with IgA-dominant immune deposits affecting small vessels (capillaries, venules, arterioles) | |
| Essential cryoglobulinemic vasculitis | Vasculitis with cryoglobulin immune deposits affecting small vessels (capillaries, venules, arterioles) and associated with cryoglobulins in serum | |
| Cutaneous leukocytoclastic angiitis | Isolated cutaneous leukocytoclastic angiitis without systemic vasculitis | |
* Large arteries include the aorta and the largest branches directed toward major body regions (e.g., the extremities, head and neck). Medium-sized arteries are the main visceral arteries (e.g., renal, hepatic, coronary, mesenteric). Small arteries are distal arterial radicles that connect with arterioles. Note that some small and large vessel vasculitides may involve medium-sized arteries, but large and medium-sized vessel vasculitides do not involve vessels smaller than arteries.
‡ Strongly associated with antineutrophil cytoplasm antibodies.
The ACR classification criteria and the CHCC definitions were not designed as diagnostic criteria. Indeed, the positive predictive value of the ACR classification criteria can be as low as 29% in clinical practice.11 Although attempts have been made to develop diagnostic criteria based on the CHCC, these Sorensen criteria have not been validated.12 Indeed, believing the ACR criteria for AGPA, PAN, and some other vasculitides, and the CHCC definitions for GPA, MPA, and PAN to no longer be fit for purpose, EULAR convened an expert consensus group to consider re-evaluating definitions, classifications, and diagnostic criteria in systemic vasculitis. Seventeen points to consider were formulated before the development of classification criteria and definitions in the systemic vasculitides that related to biopsy, laboratory testing, diagnostic radiology, nosology, definitions, and the research agenda.13 Not all of the points were relevant to AAV. Those that were relevant included the following:
• Histology point 1: Although histology is fundamental to the diagnosis of vasculitis and exclusion of its mimics, biopsy of affected organs is not always possible, and yields vary significantly according to conditions and target organs.
• Laboratory testing point 4: ANCA testing plays an important diagnostic role in suspected small vessel vasculitis.
• Diagnostic radiology point 10: Computed tomography (CT) and magnetic resonance imaging (MRI) may be useful in diagnosing ear, nose, and throat (ENT) involvement associated with GPA/AGPA.
• Nosology points 12 to 14: The nomenclature in use for distinguishing between “disease definitions,” “classification,” and “diagnostic” criteria is confusing and should be clarified wherever possible. Nosology of different forms of vasculitis should be a reflection of their etiopathogenesis, wherever this has been determined. In the absence of this, definition must rely on a clear, accurate description of the salient features of the condition. The use of eponyms should be reviewed if a more rational approach to nomenclature can be developed, based on etiopathogenesis, but their retention is necessary at present to avoid confusion.
• Definitions point 15: Age is worthy of inclusion in the definitions of some forms of vasculitis, but its role should not be overstated.
• Research agenda points 16 and 17: Future criteria initiatives should include all forms of vasculitis, providing definitions of less common syndromes not covered by CHCC. The development of a classification tree will provide the foundation for future criteria.
Epidemiology
Determining the incidence and prevalence of AAV is challenging given the uncommon occurrence of the disorders, difficulties in case ascertainment, the slow evolution of classification criteria and definitions fit for epidemiologic purposes, and the clinicopathologic overlaps that occur between the component diseases designated ANCA-associated diseases (GPA, MPA, AGPA) and their limited variants, including RLV. Most studies have been carried out in populations of European ancestry using data that were collected retrospectively. Despite these difficulties, collective studies suggest that the AAVs have an incidence of around 10 to 20 per million and prevalence rates that have been increasing over the past two decades (summarized in Reference 14). The peak age of onset is 65 to 74 years; the disease is very rare in childhood, and most studies suggest a slightly higher occurrence in men than in women (1.5 : 1.0).
Within the spectrum of ANCA-associated disease are interesting geographic and population differences in the relative incidence of GPA versus MPA or AGPA, or between MPO-ANCA and PR3-ANCA positivity. In populations of European ancestry, GPA appears to have an incidence of around 2 to 10 per million, depending on the geographic location, with the higher 8 to 10 per million incidence being reported in more northerly countries and lower incidences of 3 to 6.6 per million in Greece and Spain.15–17 A similar inverse relationship between GPA and MPA has been observed in the southern hemisphere,18 and a possible link to ultraviolet exposure has been proposed.19 However, in a Japanese population studied for occurrence of primary renal vasculitis, more than 90% had MPO-ANCA, but PR3-ANCA was not observed, and neither GPA nor AGPA was diagnosed clinically.20 In China, MPA also seems to be more common than GPA10; high rates of MPA have also been reported in a Peruvian population and in Kuwait, where the incidence of MPA was reported as 24 per million.21,22
Considering epidemiologic studies for GPA in greater detail, studies from Finland, Norway, and Sweden have suggested an increased incidence over the past two to three decades,23–25 but others from Germany and the United Kingdom and a later Swedish study have not.26–28 Overall, it is most likely that methodological factors account for reported differences, with no significant increase in incidence occurring during the past two decades. Prevalence figures for GPA have been increasing, which probably reflects improved treatment regimens. In a primary care–based population in the United Kingdom, the prevalence increased from 28.8 per million in 1990 to 64.8 per million in 2005.15 Prevalence figures are now available for several populations in Europe, the United States, and the southern hemisphere over various time periods (summarized in Reference 14).
Epidemiologic studies in MPA have variously shown an increased incidence or not over the past two decades. It is possible that earlier suggestions of an increased incidence reflected increasing awareness of MPA and its differentiation from PAN and GPA. An interesting increase in cases of MPA in Japan followed the Kobe earthquake in 1995.29
AGPA is the least common of the AAVs, with an incidence around 1.0 to 3.0 per million, although this increases in patients with asthma to 34.6 per million person-years.30 AGPA affects a similar age of population as GPA and MPA, but its occurrence is more common in women than in men. An association has been noted between the development of AGPA and the administration of leukotriene inhibitors or the anti-immunoglobulin (Ig)E monoclonal antibody omalizumab, possibly due to unmasking of previously unrecognized disease with reduction of corticosteroid dose.31,32
Environmental factors are relevant to AAV.33 There appears to be a higher incidence of GPA in rural as compared with urban areas.34 No clear seasonal variation trends have been identified. Infection as a trigger for autoimmune disease in general, and for AAV in particular, is often hypothesized. The closest association is between Staphylococcus aureus and GPA, with nasal carriage of S. aureus being linked to a higher incidence of disease relapse.35 Silica exposure has been linked to AAV in several studies, including a case-control study in the United States.36 Exposure to several drugs, including propylthiouracil, has also been associated with AAV.37 Cocaine abuse has been linked to a midline destructive granulomatous disease that mimics GPA.38
Genetics
Evidence of a heritable risk for AAV comes from recent studies suggesting a modestly increased risk of disease in first-degree relatives,39 similar to that found in rheumatoid arthritis (RA). Children of patients with GPA have an increased risk of RA,40 suggesting familial clustering of genes associated with inflammatory autoimmune disease. Occasional familial cases of GPA are also reported in the literature.41,42 Given the relatively modest risk of disease in family members, it is likely that several genes contribute a small effect on disease development. Undertaking genetic studies in AAV is challenging owing to the rarity of the disease as the statistical power of genetic association studies is determined by the number of cases and controls included in the analysis. A number of candidate genes, often involving the immune response, have been identified. However, many association studies have produced inconsistent results because of the small number of cases studied. More recent studies have used larger numbers of patients with evidence of validation of results by duplication in separate cohorts of patients. Large cohorts of patients are being assembled to allow performance of genome-wide association studies, in which thousands of genes across the genome are compared.
Consistent genetic associations with multiple autoimmune conditions have been restricted to three gene regions: human leukocyte antigen (HLA) class II region, CTLA, and the PTPN22 gene. HLA genes are exceedingly polymorphic, and small studies suggest associations between GPA and HLA compared with healthy controls. However, most of these studies are of poor reliability because of their small size and lack of replication in independent cohorts. Recently, a larger study of 150 German patients with GPA identified an association with the HLA-DPB1*0401 allele. In contrast, the *0301 allele frequency was significantly decreased.43 The extended haplotype DPB1*0401/RXB03 showed stronger association, suggesting that this genomic region confers significant risk for development of GPA. Among other functions, the retinoid X receptor β protein forms heterodimers with vitamin D receptors. Vitamin D receptor is important for the effects of the active vitamin D metabolite 1,25-dihydroxycholecalciferol. Active vitamin D has potent immunomodulatory properties, which include inhibition of cytokine transcription and differentiation of T cells toward a T regulatory (Treg) cell phenotype.44 This association has been replicated in a larger cohort of 282 GPA patients.45 The association with *0401 was present only in ANCA-positive GPA patients. Other studies have shown associations with HLA-DR. A relatively large study (304 AAV patients and 9872 controls) has shown associations with HLA-DR4,46 and another showed that HLA-DRB1*04 was increased in frequency in GPA patients with end-stage renal failure.47
Different HLA associations have been found in AGPA with disease associating with HLA-DRB4 alleles, while HLA-DRB3 afforded disease protection.48 No association of AGPA with HLA-DPB has been described.49 Different HLA associations suggest that AGPA and GPA may be different disease entities despite many similarities. This is supported by observations that showed association of the extended IL-10.2 haplotype with ANCA-negative AGPA but not GPA.49
Cytotoxic T lymphocyte–associated antigen 4 (CTLA4) is expressed mainly on activated CD4+ T cells and inhibits T cell function. Polymorphisms in the CTLA4 gene have been associated with several autoimmune diseases.50 Several studies have shown associations with polymorphisms in the CTLA4 gene. The most widely implicated CTLA4 polymorphism for risk of autoimmunity is the +49 single nucleotide polymorphism (SNP) (alanine-to-threonine substitution in the leader peptide), which appears to affect cell surface expression of CTLA4 in response to T cell activation.51,52 The CT60 SNP appears to affect the expression of soluble CTLA4 and to alter the signaling threshold of CD4+ T cells.53 Both of these SNPs have been associated with disease in patients with AAV.54–57
PTPN22 encodes the lymphoid tyrosine phosphatase LYP, which forms a complex with the kinase Csk and is a critical negative regulator of signaling through the T cell receptor. The R620W variation, which has been associated with autoimmunity, disrupts the interaction between Lck and LYP, leading to reduced phosphorylation of LYP, which ultimately contributes to gain-of-function inhibition of T cell signaling.58 Two studies have shown an association of the PTPN22 620W allele with AAV,57,59 suggesting that this is likely to contribute to the risk of AAV, as in other autoantibody-associated autoimmune diseases.
Numerous other candidate genes have been investigated in patients with AAV. Alpha1 antitrypsin (A1AT) is the major inhibitor of PR3. The A1AT gene is highly polymorphic. The deficiency alleles S and Z are increased in patients with AAV.60–63 However, most individuals with A1AT homozygous for the Z allele do not develop AAV.64
PR3 is expressed on the membrane of neutrophils and appears to be genetically determined.65 Patients with GPA tend to have higher percentages of neutrophils expressing PR3 than healthy controls,66 particularly those with relapsing disease.67 However, increased expression does not appear related to 564 promoter polymorphism of the PR3 gene.68,69 The percentage of neutrophils expressing PR3 may be influenced by HLA antigens, although the mechanism of this is unclear. In one study, a group of 34 HLA antigens was found to predict 64% of the variability in PR3 membrane expression.70 This study requires replication in an independent cohort of patients. Less support has been found for a genetic association affecting the expression of MPO. A recent meta-analysis showed no association with the functional promoter polymorphism (G-463A) in the MPO gene.71
Clinical Features
Microscopic Polyangiitis
The first descriptions of MPA were provided in 1948 by Davson, Ball, and Platt, who described an illness that they termed microscopic polyarteritis, distinguishing it from polyarteritis nodosa by its marked segmental necrotizing glomerulonephritis and greater propensity to involve small vessels.72
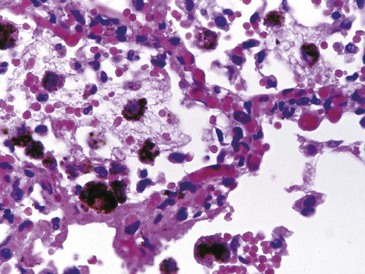
The pathology of MPA is of a fibrinoid necrotizing vasculitis with few or no immune deposits that primarily affects small vessels such as capillaries, arterioles, or venules, although spread to include small and medium-sized arteries may occur. A focal segmental necrotizing glomerulonephritis is very common and may progress into a full blown crescentic glomerulonephritis.73 However, in view of the sometimes indolent nature of the disease, evidence of chronic damage with obsolete glomeruli and tubulointerstitial fibrosis may be present at the time of the first diagnostic kidney biopsy.74 Examination of the kidney by immunohistology or electron microscopy reveals few or no immune deposits, although careful inspection may indicate that some complement fragments are present.75 Within the lungs, a pulmonary capillaritis may develop, and, following rupture of capillaries, blood can spill into alveolar spaces and thrombosis may occur within capillaries themselves. Marked neutrophilic infiltration of the alveolar wall is usually seen; this ultimately undergoes fibrinoid necrosis. Type II epithelial cell hyperplasia and lymphoplasmacytic infiltration can develop.
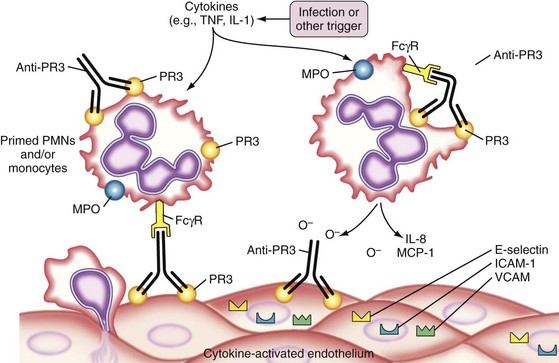
The pathophysiologic understanding of MPA advanced considerably after the strong association between MPA and ANCA was recognized, particularly ANCA directed toward myeloperoxidase (MPO).2,76 A small percentage of patients have ANCA directed toward proteinase-3 (PR3). ANCAs are believed to bind to their target antigen on the surface of primed neutrophils, leading to further neutrophil activation with release of proinflammatory granule contents and reactive oxygen species, as well as increasing adherence and damage to endothelial cells (Figure 89-2) (reviewed in Reference 77). Studies in mouse and rat species have provided direct evidence that anti-MPO antibodies induce systemic vasculitis, including necrotizing glomerulonephritis,78,79 and can promote neutrophil-endothelial cell interactions in vivo.80,81
The clinical features of MPA have been recognized for many years82–85 and are summarized in Table 89-2.The most commonly affected organs are the kidneys and the lungs. Symptoms can involve the ears, nose, or throat, but distinction from GPA then needs to be considered. The presentation can be insidious with symptoms of deteriorating renal function on the background of mild features that can be attributed to a low-grade vasculitis; alternatively, it may be severe and acute with rapidly progressive glomerulonephritis and pulmonary hemorrhage, presenting as a pulmonary renal syndrome; or it may appear to be limited to the kidney, sometimes being described as renal-limited vasculitis.
Table 89-2 Principal Clinical Features of Microscopic Polyangiitis
| Clinical Feature | Percentage* |
|---|---|
| Constitutional symptoms | 76-79 |
| Fever | 50-72 |
| Renal disease | 100 |
| Arthralgias | 28-65 |
| Purpura | 40-44 |
| Pulmonary disease (hemorrhage, infiltrates, effusion) | 50 |
| Neurologic disease (central, peripheral) | 28 |
| Ear, nose, throat involvement | 30 |
* Percentage of a population totaling 150 patients from four studies.82–85
Renal manifestations include microscopic hematuria, an abnormal renal sediment with red cell casts, proteinuria that is not usually of nephrotoxic proportions (i.e., is less than 3.5 g per 24 hours), and variable loss of kidney function. The presence of red cell casts is always indicative of an active glomerulonephritis, and the presence of red cells in a patient with long-standing disease may be due to active disease or to damage in the absence of disease activity, or indeed to other conditions affecting the lower urinary tract, including cystitis or bladder malignancy, as a result of cyclophosphamide therapy.86 Loss of renal function can be very rapid, occurring over days or weeks. Dialysis may be required, but some recovery of renal function may occur following treatment. In patients who remain on dialysis, the likelihood of relapse is less than before dialysis, so the need for immunosuppression may be less.87 Although kidney involvement is present in most patients, it is not invariable.
Lung involvement occurs in up to a third of patients.82–85 Clinical features include cough, dyspnea, pleurisy, and hemoptysis. Onset may be insidious or acute and severe. Widespread pulmonary hemorrhage can occur without overt hemoptysis. Chest radiograph findings may be patchy or diffuse, reflecting alveolar infiltrates. Repeated episodes of lung hemorrhage can lead to pulmonary fibrosis.
Granulomatosis with Polyangiitis
GPA is a granulomatous disorder, often associated with fibrinoid necrotizing vasculitis, which was first described in 1931 by Klinger,88 with pathologic refinement added to the description by Wegener in 1936.89
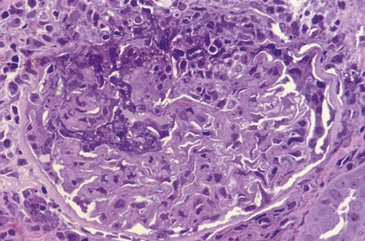
The pathology of GPA comprises granulomas and necrosis, as well as the vasculitis, which is similar to that occurring in MPA.73 Pathologic biopsy material is taken more frequently from the nasal mucosa, lung, skin, or kidney. All features are not usually present in any one biopsy specimen, given the small sample of material that may be available, so findings may be compatible with the diagnosis but not diagnostic. In the lung, open biopsies generally are more likely to be diagnostic than transbronchial biopsies.90,91 Renal pathology is similar between MPA and GPA, and granulomas are rarely seen in the kidney (Figure 89-3); sometimes intense periglomerular leukocytic infiltration has the appearance of pseudogranulomas.92
The pathophysiology of GPA, as with MPA, is believed to be inherently autoimmune93 and driven by PR3-ANCA in a manner very similar to that proposed for MPO-ANCA,77 with PR3-ANCA being highly specific for GPA.76,94 However, a good animal model is not available for anti-PR3 antibodies, so direct evidence for a role in development of vasculitis, or indeed in granuloma formation, is not available as with anti-MPO antibodies; this may reflect differences in expression of PR3 on murine neutrophils. However, the ex vivo effects of MPO-ANCA and PR3-ANCA on human neutrophils are very similar (reviewed in Reference 77). B cells and T cells are also acknowledged to play important roles in the AAV diseases, with recent demonstration of responsiveness to anti–B cell therapies placing increased interest in the role that B cells play.95,96 T cell subset abnormalities have repeatedly been described in MPA and GPA, but the nature of the factors responsible for these changes has not been defined.
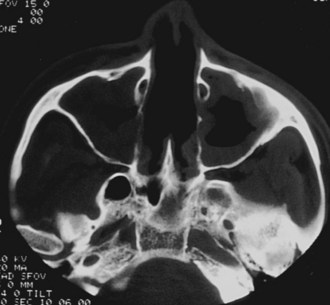
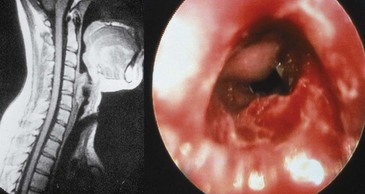
The clinical features of GPA are driven by a predilection for the upper and lower respiratory tracts, as well as for the kidneys. GPA may occur as a disease limited to the respiratory tract without evidence of systemic involvement, when it is referred to as limited GPA; such presentation is usually as a granulomatous disorder without vasculitic features. In a recent study, 10% of patients evolved to develop generalized disease within a median time of 6 years.97 Upper airway disease is the most common presenting feature of GPA, occurring in more than 70% and eventually being present in more than 90% of patients.90,98 Serous otitis media and conductive and sensorineural hearing loss may occur; vertigo is rare. Nasal involvement causes mucosal swelling with obstruction, crusting, septal perforations, serosanguineous discharge, and epistaxis; a saddle nose deformity due to collapse of the cartilaginous portion of the nasal septum may develop (Figure 89-4). Sinusitis is common and will occur in more than 80% at some point during the illness (Figure 89-5)99; bony erosion may develop and can be detected better using CT scanning of the sinuses than by plain radiography. S. aureus may infect sinuses and is commonly carried on the diseased nasal mucosa, where it may be one factor contributing to disease relapse.35 Laryngotracheal disease may cause hoarseness but may also progress to severe stridor and upper airway obstruction, usually following subglottic stenosis (Figure 89-6). Direct laryngoscopy may reveal an ulcerated friable mucosa, and tracheal tomograms, CT, or MRI can help to further delineate the extent of the stenosis. In occasional patients, subglottic stenosis can precede the development of other features of the disease by years; in others, subglottic stenosis may develop despite apparently effective control of the disease.
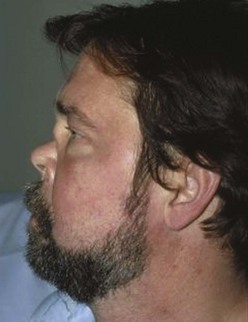
Figure 89-4 Saddle-nose deformity in a patient with granulomatosis with polyangiitis.
(Courtesy Dr. G. Hoffman.)
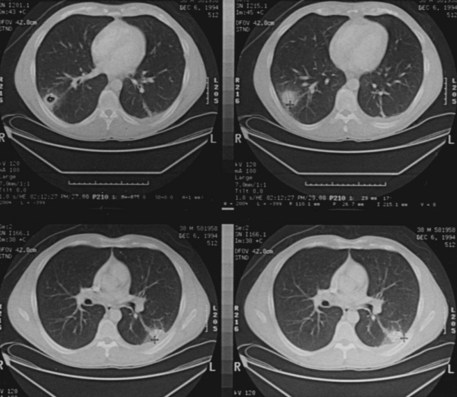
Pulmonary involvement will affect around 90% of patients at some point during the course of disease.90 Symptoms are similar to those associated with MPA, although some patients may have asymptomatic disease that is detected only after imaging. In addition to the capillaritis (Figure 89-7) and vasculitis that occur in MPA, patients with GPA are burdened with granulomatous disease that causes development of nodules of chronic granulation tissue that can cavitate centrally.100 The most common radiographic findings are pulmonary infiltrates and nodules (Figure 89-8). Infiltrates may be fleeting in nature but may be extensive, particularly when associated with severe pulmonary hemorrhage. Small nodules that are not apparent radiologically may be detected by CT scanning. Imaging may also demonstrate pleural effusion and mediastinal or hilar lymph node enlargement. Infection may be superimposed on the disease background; it is important to exclude this by culture and by bronchoscopy if necessary.101 If pulmonary hemorrhage is recent, the carbon monoxide diffusion capacity may be reduced if a patient is well enough to allow the test to be undertaken. Depending on the progress of the disease, pulmonary function tests may show an obstructive or a restrictive component, particularly if fibrosis has developed following repeated episodes of hemorrhage or cyclophosphamide-induced pneumonitis.102
The features of renal involvement in systemic GPA are similar clinically and pathologically to those in MPA. Renal involvement probably occurs in around 80% of patients at some time during the course of the illness,90,99 although determining the precise frequency overall is difficult. However, it should never be assumed that limited disease excludes the future development of renal and systemic disease because, as already noted, a proportion of patients will develop systemic disease at some point.97 The lower urinary tract may also be affected with necrotizing vasculitis of the bladder, necrotizing urethritis, orchitis, epididymitis, prostatitis, and penile necrosis (reviewed in Reference 103). Urinary obstruction may develop, particularly with involvement of the ureters. Cystoscopy should be undertaken if unexplained persistent hematuria is present, to exclude bladder malignancy or other complications.
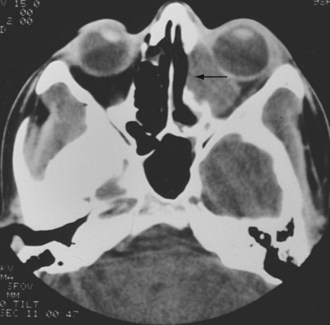
The eye may be affected in several ways during the course of GPA in 28% to 58% of patients.104,105 Keratitis, conjunctivitis, episcleritis, scleritis, uveitis, retrobulbar granulomatous disease with proptosis, ocular palsies, lacrimal duct obstruction, optic neuritis, and retinal vascular occlusion may all occur.104–106 Proptosis and optic neuritis are feared because they are particularly likely to lead to visual loss. Both CT and MRI may be helpful in defining retrobulbar disease (Figure 89-9). In patients treated with high doses of corticosteroids, cataracts may occur as a complication.
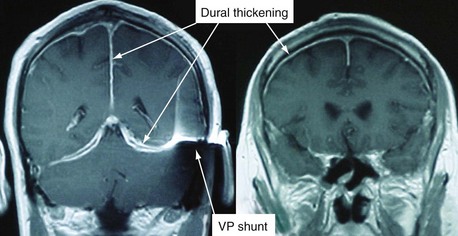
Both the peripheral and the central neurologic system can be affected in GPA.107 Peripheral neuropathy may manifest as a mononeuritis multiplex or, less commonly, as a distal and symmetric polyneuropathy. Nerve conduction studies may be helpful in verifying the presence and extent of neuropathy, and biopsy of a nerve, usually the sural nerve, may establish the presence of vasculitis. Central nervous system disease occurs in a minority of patients—around 10%—but can be severe, particularly when a cerebral vasculitis is present.108 Chronic pachymeningitis, cerebral hemorrhage and thrombosis, pituitary involvement, cerebral nerve involvement, brain stem lesions, spinal cord involvement, and subarachnoid or subdural hemorrhage may occur. Substantiating the presence of cerebral vasculitis may be difficult because small vessels are affected and angiography is of little utility. However, CT and MRI may help to define infarctions, hemorrhages, mass lesions, meningeal involvement, and white matter changes (Figure 89-10). Lumbar puncture may be necessary if subarachnoid hemorrhage is suspected or to exclude the presence of meningeal infection.
Cardiac involvement through coronary artery vasculitis and involvement of the myocardium by granulomatous disease may occur, and magnetic resonance angiography and contrast-enhanced MRI may be useful in its detection.109 Increased risk of cardiovascular disease is noted in patients with AAV, including GPA.110,111
Symptomatic involvement of the gastrointestinal tract is not usually a major feature of GPA, or indeed of MPA. However, abdominal pain, bleeding, and diarrhea can occur as a result of the disease itself or through the effects of treatment, for example, corticosteroids can cause peptic ulceration and mycophenolate mofetil may cause diarrhea. The vasculitic process itself may lead to ulcerations, or even perforations, in the small or large intestine. A number of unusual presentations include involvement of the tongue and salivary glands (parotic, sublingual, or submandibular),112 pancreatic involvement that may mimic pancreatic cancer,113 and cholecystitis and hepatic granulomatous disease that can cause liver failure.114 Splenic involvement with vasculitis, granulomas, and necrosis is present in many patients in older autopsy series.115
Allergic Granulomatosis with Polyangiitis
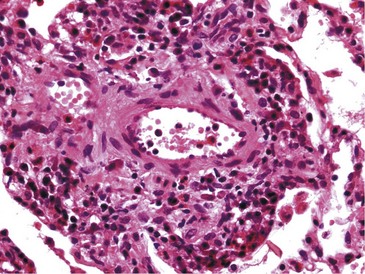
This syndrome was described by Churg and Strauss in 1951.116 It has three salient histopathologic features, namely, necrotizing vasculitis, tissue infiltration by eosinophils, and extravascular granulomas (Figure 89-11). To improve recognition of this disorder, Lanham suggested that diagnosis be based on clinical features comprising asthma, peak eosinophil count greater than 1500 cells/mL, and systemic vasculitis involving two or more organs.117,118 None of these clinical or pathologic features is entirely specific for AGPA, and they may not all be present or detectable concurrently.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


