116 Amyloidosis
The term amyloidosis includes diseases that have in common the extracellular deposition of insoluble fibrillar proteins in tissues and organs. These diseases are a subset of a growing group of disorders recognized to be caused by misfolding of proteins; they include Alzheimer’s and other neurodegenerative diseases, prion diseases, serpinopathies, some of the cystic fibroses, and others. A unifying feature of the amyloidoses is that the deposits share a common β-pleated sheet structural conformation that confers unique staining properties. The name “amyloid” is attributed to the pathologist Virchow, who in 1854 thought such deposits in autopsy livers were cellulose because of their peculiar staining reaction with iodine and sulfuric acid.1 In the 20th century, amyloid was found to be a proteinaceous fibrillar deposit in tissues.2 Biochemical characterization of fibril proteins from clinical cases proved the amyloidoses to be diverse diseases, many with a potentially fatal outcome owing to progressive deposition of amyloid fibrils in major organs. A growing number of treatments are available to target the source of the abnormal protein and, for some types, to inhibit the amyloidogenic protein misfolding process.
Classification and Epidemiology
Amyloid diseases are defined by the biochemical nature of the protein in the deposited fibril. The proteins are diverse and are unrelated in primary amino acid sequence, resulting in classification of amyloid diseases according to whether they are systemic or localized, acquired or inherited; their recognized clinical patterns also determine how they are classified (Table 116-1).3 Each amyloid disease has a shorthand nomenclature, expressed as A for amyloidosis and an abbreviation for the biochemical nature of the fibrils; for example, AL is amyloid of immunoglobulin light chain origin. The discussion in this chapter is limited to the systemic amyloidoses because these are the diseases that can involve the joints and are potentially confused with autoimmune rheumatologic disorders.
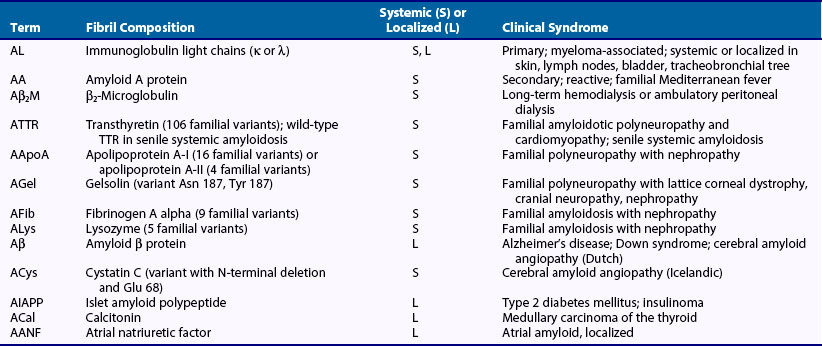
The acquired systemic amyloidoses are AL (immunoglobulin light chain, or primary), AA (reactive, secondary), and Aβ2M (β2-microglobulin, dialysis-associated) types. The AL type is most common, although epidemiologic data are limited. One study based on National Center for Health Statistics data estimated the incidence as 4.5 per 100,000.4 AL amyloidosis usually manifests after age 40 years and is associated with rapid progression, multisystem involvement, and short survival. AA amyloidosis is rare, occurring in less than 1% of patients with chronic inflammatory diseases in the United States and Europe, but it is more common in Turkey and the Middle East, where it occurs in association with familial Mediterranean fever.5 As treatments for chronic inflammation and infection improve, the origin of AA amyloidosis appears to be shifting toward rare hereditary periodic fever syndromes or hematologic disorders associated with production of inflammatory cytokines such as Castleman’s disease; workup should include genetic testing and imaging to screen for these conditions.6 The onset of AA amyloidosis is variable and can occur as soon as 1 year after the onset of an inflammatory disease or many years later, often corresponding to the degree of inflammation. It is the only type of amyloidosis that occurs in children, such as children with juvenile rheumatoid arthritis.7 Aβ2M amyloidosis is a chronic rheumatologic complication that occurs in a few patients on long-term dialysis and is related to a high concentration of β2-microglobulin.8
The inherited amyloidoses are rare autosomal dominant diseases in which a variant plasma protein forms amyloid deposits beginning in midlife.9 The most common form is caused by variant transthyretins (TTRs), of which more than 100 types are known to be associated with amyloidosis.10 One variant, with a substitution of isoleucine for valine at position 122 (V122I), has a carrier frequency that may be 4% in the black population and is associated with late-onset cardiac amyloidosis. Among a large cohort of African-Americans over age 65, the frequency of congestive heart failure and mortality was higher among those carrying the V122I gene than in age, gender, and ethnically matched controls.11 In an amyloidosis referral population, African-Americans over age 60 who presented with amyloid cardiomyopathy were twice as likely to have TTR amyloidosis (ATTR) due to V122I as to have the more common AL type of amyloidosis.12 Even wild-type TTRs can form fibrils, leading to senile systemic amyloidosis, which predominantly affects the heart, in older patients.13
Pathology and Pathogenesis of Amyloid Fibril Formation
Pathologic Features
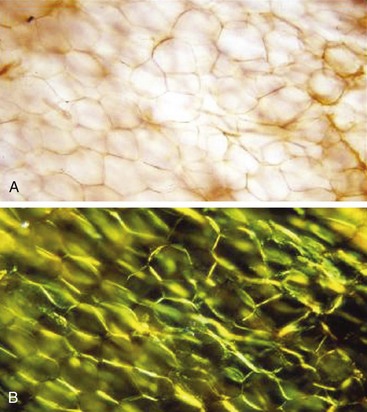
All amyloid deposits stain with Congo red dye and exhibit a unique green birefringence by polarized light microscopy,14 although the deposits also can be recognized on routine hematoxylin and eosin–stained sections. By electron microscopy, amyloid fibrils are 8 to 10 nm wide and of varying lengths, with a 2.5- to 3.5-nm filamentous subunit arranged along the long axis of the fibril in a slow twist.15 Typing of amyloid deposits can be done with conventional immunohistochemical staining. False-positive results can occur, however, owing to the presence of nonamyloid serum proteins, and immunoelectron microscopy can provide more definitive immunologic identification of the protein in the fibril, or the composition of extracted fibrils can be identified by mass spectrometric proteomic analysis.16,17
Pathogenesis of Amyloid Fibril Formation
The exact mechanism of fibril formation is unknown and may differ among the various types of amyloid.18 Studies suggest a common underlying mechanism, however, in which a partially unfolded protein intermediate forms multimers and then higher-order polymers. Factors that contribute to fibrillogenesis include variant or unstable protein structure, extensive β-conformation of the precursor protein, proteolytic processing of the precursor protein, association with components of the serum or extracellular matrix (e.g., serum amyloid P [SAP] component, amyloid enhancing factor, apolipoprotein E, glycosaminoglycans), and physical properties such as pH of the tissue site.
AL amyloidosis is a plasma cell dyscrasia with an excess of clonal plasma cells in the bone marrow; it can occur in isolation or in combination with multiple myeloma. Similar cytogenetic changes have been identified in these plasma cell diseases, suggesting that they may have a common molecular pathogenesis.19,20 By two-dimensional gel electrophoresis and mass spectrometry, it can be seen that the amyloid fibril deposits are composed of intact 23-kD monoclonal immunoglobulin light chains and C-terminal truncated fragments.16 Although all κ and λ light chain subtypes have been identified in amyloid fibrils, λ subtypes predominate, and the λ 6 subtype seems to have unique structural properties that predispose it to fibril formation,21 often in the kidney.22 AL amyloidosis is usually a rapidly progressive disease with amyloid deposits in multiple tissue sites.
The AA type of amyloidosis is a complication of severe, long-standing inflammation, as occurs in chronic inflammatory disorders or infections. AA amyloid fibrils usually are composed of an 8-kD, 76 amino acid amino-terminal portion of the 12-kD precursor, serum amyloid A (SAA).23 SAA is a polymorphic protein encoded by a family of SAA genes, which are acute phase apoproteins synthesized in the liver and transported by a high-density lipoprotein, HDL3, in the plasma.24 An underlying inflammatory disease of several years’ duration causing an elevated SAA usually precedes fibril formation, although infection can produce AA deposition more quickly. AA fibril formation can be accelerated by an amyloid enhancing factor present in high concentration in the spleen (which may be early SAA aggregates or deposits), by basement membrane heparan sulfate proteoglycan, or by seeding with AA or heterologous fibrils.25,26
Factors related to β2-microglobulin fibril formation are under investigation. The high prevalence of Aβ2M disease in patients undergoing long-term dialysis argues against an amyloidogenic variant β2-microglobulin molecule. The permeability of dialysis membranes may be a factor because the molecular weight of β2-microglobulin is 11.8 kD—above the porosity of standard membranes. It has been hypothesized that dialysis membranes may be bioincompatible and may induce proinflammatory mediators that stimulate β2-microglobulin and contribute to fibril formation.27
In ATTR (also called familial amyloidotic polyneuropathy) and all other forms of familial amyloidosis, inherited mutations or polymorphisms in the genes encoding large serum proteins produce amyloid-prone variants. The process of fibrillogenesis has been best studied for TTR, in which variant TTR molecules seem to be prone to dissociation from stable tetramers and to unfolding, leading to misfolding, polymerization, and fibril formation.28 The role of aging is intriguing because patients with variant proteins do not have clinically apparent disease until midlife or later, despite the lifelong presence of the abnormal protein.29 Further evidence of an age-related trigger is that senile cardiac amyloidosis, caused by the deposition of fibrils derived from normal TTR, is exclusively a disease of elderly individuals.13
Diagnosis
A tissue biopsy specimen showing amyloid fibrils is necessary for the diagnosis of amyloidosis (Figure 116-1). The least invasive biopsy is the abdominal fat aspirate, which is positive in 80% to 90% of patients with AL or ATTR amyloidosis and in 60% to 70% of patients with AA amyloidosis.30,31 It is easy to perform after local injection of anesthetic and has a low rate of infectious or hemorrhagic complications (Figure 116-2). If the aspirate is negative, but clinical suspicion for disease persists, a more invasive tissue biopsy should be done. Although a biopsy specimen of a clinically involved organ is recommended, almost any tissue biopsy specimen is likely to be positive if the patient has systemic amyloidosis: In a series of 100 patients with AL amyloidosis, 85% of 249 tissue biopsy specimens were positive, including all samples from the kidney, heart, and liver.32 When the diagnosis of amyloidosis is made, careful evaluation of the entire clinical picture, including manner of presentation, organ system involvement, underlying disease, and family history, should provide a clue to the type of amyloid.
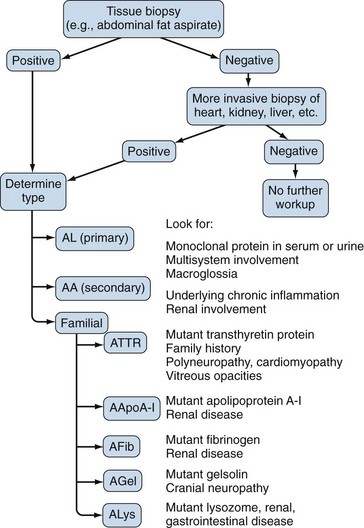
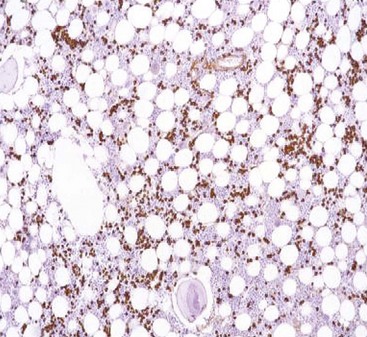
Identification of a plasma cell dyscrasia distinguishes AL from other types of amyloidosis (Figure 116-3). More than 90% of patients have a serum or urine monoclonal immunoglobulin protein or a free light chain on testing by immunofixation electrophoresis or by a recently available nephelometric assay for free light chains.33,34 In addition, the percentage of plasma cells in the bone marrow is often increased; these are monoclonal on immunohistochemical staining (Figure 116-4).35 A monoclonal serum protein by itself is not diagnostic of amyloidosis because monoclonal gammopathy of uncertain significance is common in older patients. When monoclonal gammopathy of uncertain significance is present in a patient with biopsy-proven amyloidosis, however, the AL type is strongly suspected. Immunohistochemical staining by light or electron microscopy should be done by a laboratory familiar with the techniques and able to perform appropriate controls. Mass spectrometry–based microsequencing of small amounts of protein extracted from fibril deposits ultimately may be the most reliable way to identify the components of the fibrils.16,17AA amyloidosis is suspected in patients with renal amyloidosis and a chronic inflammatory condition or infection. AL and ATTR amyloidosis must be ruled out. AA amyloidosis must be confirmed by immunohistochemical staining for AA protein.
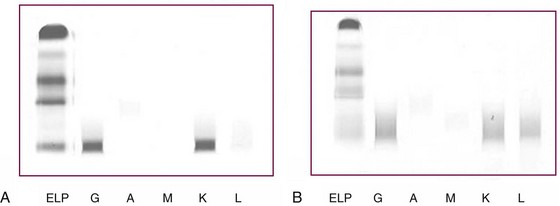
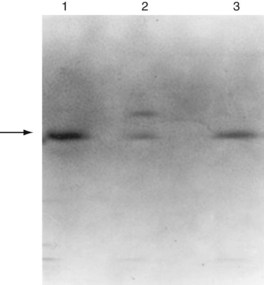
Familial amyloidosis must be excluded in every patient who does not have a plasma cell dyscrasia or the AA type of amyloidosis. Although the disease has a dominant inheritance, family history may not be apparent when the disease occurs later in life; also, some cases occur through new mutations. Variant TTR proteins usually can be detected by isoelectric focusing (Figure 116-5).36 Abnormal isoelectric focusing should prompt genetic testing to determine the precise TTR mutation. Genetic testing should be employed when screening tests fail to identify the fibril protein. With the use of polymerase chain reaction–based sequencing, abnormal fibrinogens and apolipoproteins and variant TTRs can be detected.37
A novel renal amyloid protein, leukocyte chemotactic factor 2 (LECT2), has been found in glomeruli, renal vessels, and interstitium in patients with isolated renal amyloidosis and should be considered in patients who are negative for the more common types of amyloidosis.38
Clinical Features and Treatment of the Systemic Amyloidoses
AL Amyloidosis
AL amyloidosis usually occurs in middle-aged or older individuals but also can occur in the third or fourth decade of life. It has a wide spectrum of organ system involvement, and presenting features reflect the organs most prominently affected.39,40 Initial symptoms of fatigue and weight loss are frequent, but the diagnosis is rarely made until symptoms referable to a specific organ appear.
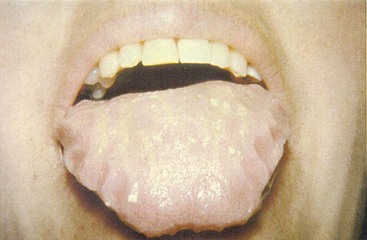
The kidneys are commonly affected; renal amyloidosis is manifested by proteinuria, sometimes massive, with edema and hypoalbuminemia. Mild renal dysfunction is frequent, but rapidly progressing renal failure is rare. Cardiac involvement, often with congestive heart failure, is a common presentation.41 The electrocardiogram may show low voltage with a pattern of myocardial infarction. The echocardiogram frequently shows concentrically thickened ventricles and a normal or mildly reduced ejection fraction. Nervous system features include peripheral sensory neuropathy, carpal tunnel syndrome, and autonomic dysfunction with gastrointestinal motility disturbances (early satiety, diarrhea, constipation) and orthostatic hypotension. Macroglossia, a classic feature pathognomonic of AL amyloidosis, is found in 10% of patients (Figure 116-6). Hepatomegaly may be massive with mild cholestatic abnormalities of liver function, although liver failure is uncommon, even when hepatomegaly is massive. The spleen is frequently involved, and functional hyposplenism may occur, even in the absence of significant splenomegaly. Cutaneous ecchymoses are common, particularly around the eyes, giving the “raccoon-eyes” sign, and appear spontaneously or when provoked by minor trauma (Figure 116-7). Other findings include nail dystrophy (Figure 116-8), alopecia, and amyloid arthropathy with thickening of synovial membranes. We have reviewed the soft tissue and joint manifestations of AL amyloidosis in a series of almost 200 patients.42 Symptoms and signs mimicking rheumatologic diseases, with arthropathy, subcutaneous tissue deposits, muscle pseudohypertrophy, adenopathy, carpal tunnel syndrome, submandibular gland enlargement, and macroglossia, occur in more than 40% of patients with AL amyloidosis.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


