13 T Lymphocytes
Overview
The evolutionary pressures that have molded the immune response and promoted a highly diverse repertoire clearly derive from infectious agents. Two different strategies exist. The more primitive innate immune response (see Chapter 18) uses a limited repertoire of nonpolymorphic receptors that recognize structural motifs that are common to many microorganisms. These include small glycolipids and lipopeptides. The alternative strategy of the evolutionarily newer adaptive immune response (see Chapter 19) relies on generating myriad different receptors that can recognize a wide array of foreign compounds from infectious agents. Whereas the innate immune response allows a rapid focused response, adaptive immunity permits a broader, albeit slower, response but critically offers the additional benefit to the host of immune memory.
T lymphocyte development constantly confronts the dilemma of combating infection without provoking a response to the host. The price for generating an increasingly varied population of antigen receptors needed to recognize a wide spectrum of pathogens is the progressive risk of producing self-reactive lymphocytes that can provoke an autoimmune diathesis. To minimize the possibility of self-reactive cells, T lymphocytes are subjected to a rigorous selection process during development in the thymus. In addition, premature activation of mature T cells is prevented by requiring two signals for activation. Finally, the tremendous expansion of T cells that occurs during the response to an infection is resolved by the active induction of cell death. The consequences of inefficient lymphocyte removal at any one of these junctures can be devastating to the health of the organism. This is vividly displayed in both humans and mice where naturally arising mutations in death receptors such as Fas result in massive accumulation of lymphocytes and autoimmune sequelae. These are discussed in more detail in Chapter 27 on cell survival and death.
The activation of T lymphocytes yields a variety of effector functions that are pivotal to combating infections. Cytolytic T cells can kill infected cells through the expression of perforin, which induces holes in cell membranes, or ligands for death receptors such as Fas or tumor necrosis factor (TNF) receptor. Production of T cell cytokines such as interferon-γ (IFN-γ) can inhibit viral replication, whereas other cytokines such as interleukin (IL)-4, IL-5, and IL-21 are critical for optimal B cell growth and immunoglobulin production.1 However, this same armamentarium, if not tightly regulated, can also precipitate damage to host tissues and provoke autoimmune responses. This is particularly apparent in situations where T cell infiltration can be observed histologically such as in the synovium of inflammatory arthritides, pancreatic islets in type 1 diabetes, and the central nervous system in multiple sclerosis. Damage in these cases need not be the direct result of recognition of target tissues by the T cells. T cells may be activated elsewhere and then migrate to the tissue and damage innocent bystander cells. T cells may also promote autoimmunity through the augmentation of B cell responses.
T Cell Development
The TCR is an 80- to 90-kD disulfide-linked heterodimer composed of a 48- to 54-kD α chain and a 37- to 42-kD β chain. An alternate TCR composed of γ and δ chains is expressed on 2% to 3% of peripheral blood T cells and is discussed later. The TCR has an extracellular ligand binding pocket and a short cytoplasmic tail that by itself cannot signal. Consequently it is noncovalently associated with as many as five invariant chains of the CD3 complex that relay information to the intracellular signaling machinery via immunoreceptor tyrosine activation motifs (ITAMs) (see later). Not surprisingly, the structure of the TCR gene is similar to what was first described for immunoglobulin genes in B cells (see details in Chapter 14). Each overcame the problem of how to encode approximately 10 million different T or B cell specificities within the human genome, which contains only 30,000 genes. To economically package this diversity, the process of gene rearrangement and splicing evolved using machinery similar to that which already existed to promote gene translocations. The β and δ chain genes of the TCR contain four segments known as the V (variable), D (diversity), J (joining), and C (constant) regions. The α and γ chains are similar but lack the J component. Each of the segments has several family members (≈ 50 to 100 V, 15 D, 6 to 60 J, and 1 to 2 C members). An orderly process occurs during TCR gene rearrangement in which a D segment is spliced adjacent to a J segment, which is subsequently spliced to a V segment.
Development of T cells occurs within a microenvironment provided by the thymic epithelial stroma. The thymic anlage is formed from embryonic ectoderm and endoderm and is then colonized by hematopoietic cells, which give rise to dendritic cells, macrophages, and developing T cells.2 The hematopoietic and epithelial components combine to form two histologically defined compartments: the cortex, which contains immature thymocytes, and the medulla, which contains mature thymocytes (Figure 13-1A). As few as 50 to 100 bone marrow–derived stem cells enter the thymus daily.
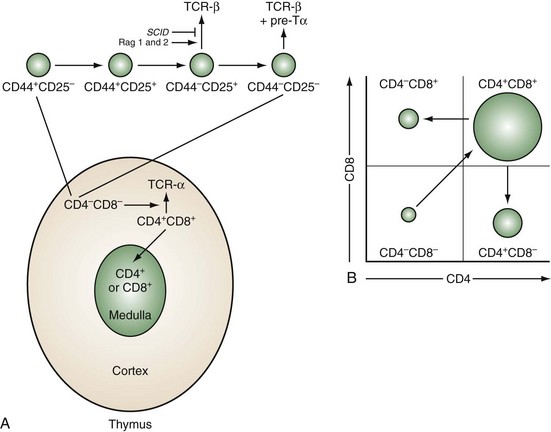
Figure 13-1 Sequence of thymocyte development. A, The earliest thymocyte precursors lack expression of CD4 and CD8 (CD4−CD8−). These can be further divided into four subpopulations based on sequential expression of CD44 and CD25. It is at the CD44−CD25+ stage that the TCR-β-chain rearranges. The SCID mutation or deficiencies of the rearrangement enzymes Rag-1 and Rag-2 result in inability to rearrange the β-chain and maturational arrest at this stage. Those thymocytes that successfully rearrange the β-chain express it associated with a surrogate α-chain known as pre-Tα. Concomitant with a proliferative burst, development can then progress to the CD4+CD8+ stage in the cortex where the TCR-α-chain rearranges and pairs with the β-chain to express a mature TCR complex. These cells then undergo thymic positive and negative selection (as diagrammed in Figure 13-3B). Successful completion of this rigorous selection process results in mature CD4+ or CD8+ T cells in the medulla, which eventually emigrate to peripheral lymphoid sites. B, Schematic two-color flow cytometry showing subpopulations of thymocytes defined by CD4 and CD8 expression in their relative proportions.
The stages of thymocyte development can be defined by the status of rearrangement and expression of the two genes that encode the α and β chains of the TCR and the expression of CD4 and CD8, proceeding in an orderly fashion from CD4−8− → CD4+8+ → CD4+8− or CD4−8+ (Figure 13-1B). CD4 and CD8 define, respectively, the helper and cytolytic subsets of mature T cells.
CD4−8− thymocytes can be further subdivided based on their expression of CD25 (the high-affinity IL-2 receptor α chain) and CD44 (the hyaluronate receptor).3 Development proceeds in this order: CD25−CD44+ → CD25+CD44+ → CD25+CD44− → CD25−CD44−. These subpopulations correspond to discrete stages of thymocyte differentiation. CD25−44+ cells express low levels of CD4, and their TCR genes are in germline configuration. These cells downregulate CD4 and upregulate CD25 to give rise to CD25+CD44+ thymocytes, which now express surface CD2 and low levels of CD3ε. At the next stage (CD25+CD44−), there is a brief burst of proliferation followed by upregulation of the recombination-activating enzymes, RAG-1 and RAG-2, and the concomitant rearrangement of the genes of the TCR β chain. A small subpopulation of T cells rearranges and expresses a second pair of TCR genes known as γ and δ. Productive TCR β-chain rearrangement results in downregulation of RAG and a second proliferative burst. Loss of CD25 then yields CD25−CD44− thymocytes.
The TCR β chain cannot be stably expressed without an α chain. Because the TCR α chain has not yet rearranged, a surrogate invariant TCR pre-α chain is disulfide linked to the β chain.4 When associated with components of the CD3 complex, this allows a low-level surface expression of a pre-TCR and progression to the next developmental stage. Failure to successfully rearrange the TCR β chain results in a developmental arrest at the transition from CD25+CD44− to CD25−CD44−. This occurs in RAG-deficient mice, as well as in mice and humans with SCID.5
A number of signaling molecules are required for early T cell development (Figure 13-2). The Ikaros gene encodes a family of transcription factors required for the development of cells of lymphoid origin. Notch-1, a molecule known to regulate cell fate decisions, is also required at the earliest stage of T cell lineage development.6 Cytokines including IL-7 promote the survival and expansion of the earliest thymocytes. In mice deficient for IL-7, its receptor components IL-7Rα or γc, or the cytokine receptor–associated signaling molecule JAK-3, thymocyte development is inhibited at the CD25−CD44+ stage. In humans, mutations in γc or JAK-3 result in the most frequent form of SCID.7 Pre-TCR signaling is required for the CD25+CD44− → CD25−CD44− transition. Thus loss of signaling components including Lck, SLP-76, and LAT-1 results in a block at this stage of T cell development. TCR signals are also required for differentiation of CD4+CD8+ to CD4+ or CD8+ cells. Humans deficient in ZAP-70 (see later) have CD4+ but not CD8+ T cells in the thymus and periphery.8
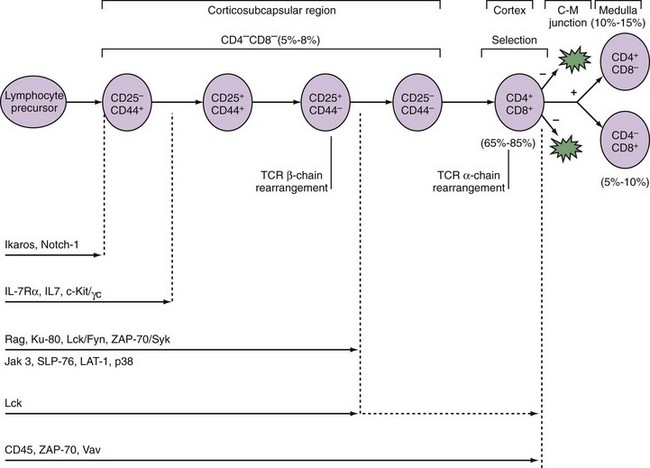
CD25−CD44− cells upregulate expression of CD4 and CD8 to become CD4+8+. It is as a CD4+CD8+ thymocyte that the α chain of the TCR rearranges. Unlike the β chain, allelic exclusion of the α chain is not apparent. Rearrangement of the α chain can occur simultaneously on both chromosomes, and if one attempt is unsuccessful, repeat rearrangements to other Vα segments are possible. Reports exist of dual TCR expression by up to 30% of mature T cells in which the same T cell expresses different α chains paired with the same β chain.9 However, in most cases of dual TCR α chains, one is downregulated during positive selection by Lck and Cbl, through ubiquitination, endocytosis, and degradation.
Although the structure of immunoglobulin and TCR are quite similar, they recognize fundamentally different antigens. Immunoglobulins recognize intact antigens in isolation, either soluble or membrane bound, and are often sensitive to the tertiary structure. The TCRαβ recognizes linear stretches of antigen peptide fragments bound within the grooves of either major histocompatibility complex (MHC) class I or class II molecules (Figure 13-3A). Thymic selection molds the repertoire of emerging TCR so that they recognize peptides within the groove of self-MHC molecules, ensuring self-MHC restriction of T cell responses. The MHC structure is described in detail in Chapters 19 through 21. Pockets within the MHC groove bind particular residues along the peptide sequence of 7 to 9 amino acids for MHC class I and 9 to 15 amino acids for MHC class II molecules. As a result, depending on the particular MHC molecule, certain amino acids will make strong contact with the MHC groove while others will contact the TCR.
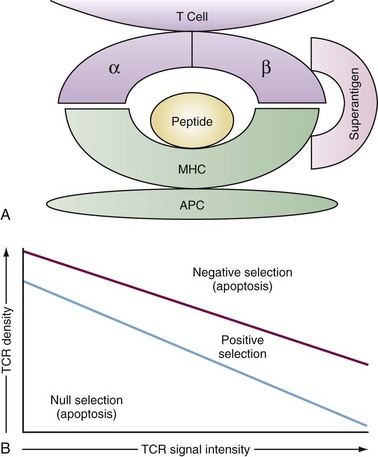
The contact between the TCR and MHC/peptide has been revealed by crystal structure to be remarkably flat, rather than a deep lock and key structure one might imagine.10 The TCR axis is tipped about 30 degrees to the long axis of the MHC class I molecule and is slightly more skewed to MHC class II. The affinity of the TCR for antigen/MHC is in the micromolar range. This is less than many antibody-antigen affinities and is several logs less than many enzyme-substrate affinities. This has led to the notion that TCR interactions with antigen/MHC are brief and that successful activation of the T cell requires multiple interactions, resulting in a cumulative signal.
Once the T cell has successfully rearranged and expressed a TCR in association with the CD3 complex, it encounters the second major hurdle in T cell development, thymic selection. Selection has two phases, positive and negative, and the outcome is based largely on the intensity of TCR signaling in response to interactions with MHC/self-peptides expressed on thymic epithelium and dendritic cells. TCR signals that are either too weak (death by neglect) or too intense (negative selection) result in elimination by apoptosis, whereas those with intermediate signaling intensity survive positive selection (Figure 13-3B). Successful positive selection at the CD4+8+ stage is coincident with upregulation of surface TCR, the activation markers CD5 and CD69, and the survival factor Bcl-2.11 T cells bearing a TCR that recognizes MHC class I maintain CD8 expression, downregulate CD4, and become CD4−8+. T cells expressing a TCR that recognizes MHC class II become CD4+8−.
An enigma for thymic selection has been how to present the myriad self-proteins to developing thymocytes so that self-reactive thymocytes are effectively eliminated by negative selection. This includes particularly those antigens with tissue or developmentally restricted expression. A solution was found with the discovery of the autoimmune regulator (AIRE) gene. AIRE is a transcription factor expressed by the medullary epithelium of the thymus that induces transcription of a wide array of organ-specific genes, such as insulin, that might otherwise be sequestered from developing thymocytes.12 This effectively creates a self-transcriptome within the thymus against which autoreactive T cells can be deleted. Gene knockout mice of AIRE and humans bearing AIRE mutations manifest various autoimmune sequelae in a syndrome known as autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED).13
Not surprisingly, a variety of signaling molecules activated by TCR engagement are important to thymic selection. Lck, the RasПRaf-1ПMEK1ПERK kinase cascade, the kinase ZAP-70, and the phosphatases CD45 and calcineurin are involved with positive selection. Among these the Ras ПERK pathway is particularly important because dominant negative variants of these molecules can disrupt positive selection. Conversely, an activator of Ras known as GRP1 assists the positive selection of thymocytes expressing weakly selecting signals. These molecules are discussed in more detail in the section on TCR signaling. By contrast, although a number of molecules may promote negative selection, among them the MAP kinases JNK and p38, there appears to be sufficient redundancy so that only rarely does elimination of any one of these molecules affect deletion of thymocytes. The few exceptions include CD40, CD40L, CD30, or the pro-apoptotic Bcl-2 family member, Bim, where preservation of at least some thymocytes bearing self-reactive TCR could be observed in mice deficient in these molecules.14–16
The survivors of these two stringent processes of TCR gene rearrangement and thymic selection represent less than 3% of total immature thymocytes. This is reflected in the presence of a high rate of cell death in developing thymocytes. This can be visualized by the measurement of DNA degradation, a hallmark of apoptosis, as shown in Figure 13-4. The survivors become either CD4+ helper or CD8+ cytolytic T cells and reside in the thymic medulla for 12 to 14 days before emigrating to the periphery. The decision to become a CD4+ versus CD8+ T cell involves further developmental signals including once again Notch-1. Notch-1 signaling is required for progression to CD8+ but not CD4+ thymocytes. This parallels the observation that long TCR interactions are required for CD4 progression, whereas shorter TCR engagement is required for CD8 progression.17
Abnormalities of Human T Cell Development
Given the vast number of events in T cell development, it is not surprising that a multiplicity of causes can underlie human T cell immunodeficiencies.18 The influence of the thymic stroma on thymocyte ontogeny is underscored in the DiGeorge anomaly in which development of the pharyngeal pouches is disrupted and the thymic rudiment fails to form. This results in the failure of normal T cell development. Less severe T cell deficiencies are associated with a failure to express MHC class I and/or class II (the “bare lymphocyte syndrome”), which are directly involved with interactions required to induce the positive selection of, respectively, CD8+ and CD4+ mature T cells.
The inability to express a number of surface molecules important in TCR and cytokine signaling also has the potential to perturb development. The failure to express TCR-CD3 components (specifically CD3γ and CD3ε), CD18, and IL-2Rγ have all been noted among patients who exhibit varying degrees of T cell deficiency or dysfunction.19 All these molecules are involved in signaling of thymocyte development and survival, and their absence clearly has the potential to alter developmental fate.
Peripheral Migration of T Cells
The migration of naïve T cells to peripheral lymphoid structures or their infiltration into other tissues requires the coordinate regulation of an array of cell adhesion molecules. T cell recirculation is essential for host surveillance and is carefully regulated by a specific array of homing receptors. Entry from the circulation to tissues occurs via two main anatomic sites: the flat endothelium of the blood vessels and specialized postcapillary venules known as high endothelial venules (HEV). A three-step model has been proposed for lymphocyte migration: rolling, adhesion, and migration.20 L-selectin expressed by naïve T cells binds via lectin domains to carbohydrate moieties of GlyCAM-1 and CD34 (collectively known as peripheral node addressin) that are expressed on endothelial cells, particularly HEV. The weak binding of CD62L to its ligand mediates a weak adhesion to the vessel wall, which, combined with the force of blood flow, results in rolling of the T cell along the endothelium. The increased cell contact assists the interaction of a second adhesion molecule on lymphocytes, the integrin LFA-1 (CD11a/CD18) with its ligands, ICAM-1 (CD54), and ICAM-2 (CD102). This results in the arrest of rolling and firm attachment. Migration into the extracellular matrix of tissues may involve additional lymphocyte cell surface molecules such as the hyaluronate receptor (CD44) or the integrin α4β7 (CD49d/β7) that binds the mucosal addressin cell adhesion molecule-1 (MAdCAM-1) on endothelium of Peyer’s patches and other endothelial cells.
Other cytokines known as chemokines may contribute to lymphocyte homing. Chemokines are structurally and functionally related to proteins bearing an affinity for heparan sulfate proteoglycan and promote migration of various cell types.21 The chemokines RANTES, MIP-1α, MIP-1β, MCP-1, and IL-8 are produced by a number of cell types including endothelium, activated T cells, and monocytes and are present at inflammatory sites such as rheumatoid synovium (see Chapter 70).
Once mature T cells have reached the peripheral lymphoid tissues of lymph node and spleen, they undergo a low level of turnover known as homeostatic proliferation in response to self-peptide/MHC complexes, IL-7, and IL-15.22 This serves to maintain the number of peripheral T cells, which can become accelerated in states of lymphopenia such as following chemotherapy or irradiation.23 Because homeostatic proliferation is driven by self-MHC/peptides, its acceleration could precipitate an autoimmune syndrome. In this regard, it is of interest that one of the standard models of autoimmunity is day 3 thymectomy, which results in lymphopenia,24 NOD diabetic mice have chronic lymphopenia that contributes to their diabetes,25 and evidence of augmented homeostatic proliferation has been suggested to occur in rheumatoid arthritis.26
Activation of T Cells
TCR and Tyrosine Kinases
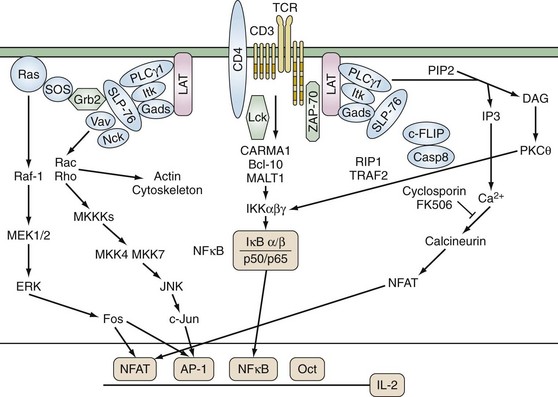
TCR αβ and γδ have short cytoplasmic domains and by themselves are unable to transduce signals. The molecules of the noncovalently associated CD3 complex couple the TCR to intracellular signaling machinery (Figure 13-4). The CD3 complex contains nonpolymorphic members known as CD3ε, CD3γ, CD3δ, and ζ and η chains that are alternatively spliced forms of the same gene and are not genetically linked to the CD3 complex. Although the functional stoichiometry of the TCR complex is not completely defined, current data indicate that each TCR heterodimer is associated with three dimers: CD3 εγ, CD3 εδ, and ζζ or ζ. CD3 ε, γ, and δ have an immunoglobulin-like extracellular domain, a transmembrane region, and a modest cytoplasmic domain, whereas ζ contains a longer cytoplasmic tail. The transmembrane domains of ζ and the CD3 chains contain a negatively charged residue that interacts with positively charged amino acids in the transmembrane domain of the TCR.
None of the proteins in the TCR complex has intrinsic enzymatic activity. Instead, the cytoplasmic domains of the invariant CD3 chains contain conserved activation domains that are required for coupling the TCR to intracellular signaling molecules. These immunoreceptor tyrosine-based activation motifs (ITAMS) contain a minimal functional consensus sequence of paired tyrosines (Y) and leucines (L): (D/E)XXYXXL(X)6-8YXXL. ITAMs are substrates for cytoplasmic protein tyrosine kinases (PTKs), and upon phosphorylation they recruit additional molecules to the TCR complex.27 Each ζ chain contains three ITAMs, whereas there is one in each of the CD3 ε, γ, and δ chains. Thus each TCR complex can contain as many as 10 ITAMs.
Activation of PTKs is one of the earliest signaling events following TCR stimulation. Four families of PTKs are known to be involved in TCR signaling: Src, Csk, Tec, and Syk. The Src family members Lck and FynT have a central role in TCR signaling and are expressed exclusively in lymphoid cells. Src PTKs contain multiple structural domains including (1) N-terminal myristylation and palmitylation sites, which allow association with the plasma membrane; (2) a Src homology (SH) 3 domain, which associates with proline-rich sequences; (3) an SH2 domain that binds phosphotyrosine-containing proteins; and (4) a carboxy-terminal negative regulatory site. Their catalytic activity is regulated by the balance between the actions of kinases and phosphatases. Activity is repressed by phosphorylation of a conserved carboxy-terminal tyrosine, and dephosphorylation by the phosphatase CD45 is critical for the initiation of TCR-mediated signal transduction. In addition, autophosphorylation of other tyrosines within the kinase domain enhances catalytic activity. Lck is physically and functionally associated with CD4 and CD8. Fifty percent to 90% of total Lck molecules are associated with CD4 and 10% to 25% with CD8. CD4 and CD8 physically associate with the TCR/CD3 complex during antigen stimulation as a result of their interaction with MHC class II and class I molecules and thus enhance TCR-mediated signals by recruiting Lck to the TCR complex. Lck phosphorylates the CD3 chains, TCRζ, ZAP-70, phospholipase C-γ1 (PLC-γ1), Vav, and Shc. Fyn binds TCRζ and CD3ε and, although its substrates are less well defined, T cells lacking Fyn have diminished response to TCR signals.28,29 Vitamin D, whose deficiency is linked to various autoimmune disorders, strongly upregulates PLC-γ1 following TCR activation and enhances signaling efficiency.30 In addition, the SH2 and SH3 domains of Src PTKs can mediate their association with, respectively, phosphotyrosine- and proline-containing molecules.
Phosphorylation of the ITAM motifs on the CD3 complex recruits the Syk kinase family member ZAP-70 by its tandem SH2 domains. ZAP-70 is expressed exclusively in T cells and is required for TCR signaling. Like the Src family PTKs, ZAP-70 is positively and negatively regulated by its phosphorylation. Phosphorylation of tyrosine 493 by Lck activates ZAP-70 kinase activity. In murine thymocytes and ex vivo T cells, inactive non-phosphorylated ZAP-70 is constitutively associated with the basally phosphorylated TCRζ chain via the SH2 domain of ZAP-70.31 TCR stimulation is required for ZAP-70 phosphorylation and activation. The recruitment of ZAP-70 to the TCR complex assists the tyrosine phosphorylation and activation of ZAP-70 by Lck. Loss-of-function hypomorphic alleles of ZAP-70 result in reduced TCR signaling and a propensity for autoimmune phenomena such as rheumatoid factor production.32
Adaptor Proteins
Two critical adaptor proteins for linking proximal and distal TCR signaling events are SH2-domain-containing leukocyte protein of 76 kDa (SLP-76) and Linker for activation of T cells (LAT) (see Figure 13-4). Loss of these adaptor proteins has profound consequences for T cell development. Mice deficient for LAT or SLP-76 manifest a block in T cell development at the CD4−8− CD25+CD44+ stage. LAT is constitutively localized to lipid rafts and, following TCR stimulation, is phosphorylated on tyrosine residues by ZAP-70. Phosphorylated LAT then recruits SH2-domain-containing proteins including PLCγ1, the p85 subunit of phosphoinositide-3 kinase, IL-2 inducible kinase (Itk), and the adaptors Grb2 and Gads. Because the SH3 domain of Gads is constitutively associated with SLP-76, this brings SLP-76 to the complex where it is phosphorylated by ZAP-70. SLP-76 contains three protein binding motifs: tyrosine phosphorylation sites, a proline-rich region, and an SH2 domain. The N terminus of SLP-76 contains tyrosine residues that associate with the SH2 domains of Vav, the adaptor Nck, and Itk. Vav is a 95-kD protein that acts as a guanine nucleotide exchange factor for the Rho/Rac/cdc42 family of small G proteins. The complex of LAT, SLP-76/Gads, PLCγ1, and associated molecules results in the full activation of PLCγ1 and activation of Ras/Rho GTPases and the actin cytoskeleton.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


