Fig. 9.1
Timing and Genetic Consequence of de novo Mutations De novo mutation may occur at any time during embryonic development, ranging from prior to fertilization to after birth. Mutations occurring prior to fertilization result in parental gonadal mosaicism; every cell in the developing embryo will contain the mutation (shown with red color). Mutations occurring during early embryonic development will result in somatic mosaicism in the developing embryo. The timing of the de novo mutation will, in part, dictate the extent to which the mutation occurs in the embryo/child. Examples for each are shown below
Timing of De Novo Mutations
As noted, de novo mutations may occur randomly and their timing affects development in the child (Fig. 9.1). In general, every cell in a person’s body carries the identical genetic content. However, the term genetic mosaicism describes the accumulation of de novo mutations resulting in different cells carrying a slightly different genetic content (ie. one cell carries de novo mutations while other cells do not) [4]. Gonadal mosaicism occurs when the germ cells (sperm and egg cells) contain de novo mutations prior to fertilization. When a sperm cell contains a de novo mutation, it is genetically different from all other cells, a phenomenon commonly associated with advanced paternal age. If this sperm cell fertilizes an egg, every cell of the offspring will contain that same mutation. These mutations are easily identified during genetic analysis, as they are present in blood samples from the child but not from either parent.
If de novo mutations occur after fertilization, either during embryonic development or after birth, the child will be mosaic (also called somatic mosaicism). In this scenario, the child (not the parent) is mosaic. As cells divide, cells with the de novo mutation give rise to mutated cells, while the other cells are normal. Ultimately, the proportion of mutant cells in the child is determined, in part, by the timing of the original mutational event. Events during early embryonic development will yield more cells in the patient that carry the mutation, while mutations that occur later in development or even after birth will result in fewer mutant cells that may be localized to a particular tissue, organ, or structure. For example, cancers arise from somatic mutations occurring after birth and are often localized to a particular tissue or organ [5, 6]. Genetic analyses that compare DNA from ‘affected’ and ‘normal’ tissues are required to identify these somatic mosaic de novo mutations.
De Novo Mutations in Orthopaedic Disorders
Mutations associated with paternal age effects are thought to arise due to replication errors during spermatogenesis. During this process, male germ cells continuously divide, and as the number of DNA replications increases, so does the risk of error and de novo mutation. Achondroplasia is one of several hallmark disorders described as having a paternal age effect; Wilhelm Weinberg in 1912 first described an increased incidence of achondroplasia among ‘last-born’ children. As observed in 1912, and again later by Lionel Penrose in 1955, the incidence of achondroplasia increases with paternal age but in an accelerated, non-linear rate [7]. Achondroplasia (Fig. 9.2) is a form of disproportionate short-limb dwarfism caused by an abnormal three-dimensional configuration of the receptor for fibroblast growth factor 3 (FGFR3) [8–10]. Achondroplasia is caused most frequently by a spontaneous missense mutation (G380R) in the protein’s transmembrane domain that is subsequently transmitted in an autosomal dominant pattern [8–10]. In addition to achondroplasia, other skeletal syndromes associated with advanced paternal age include craniofacial and skeletal dysplasias such as Apert syndrome (Fig. 9.3), Crouzon syndrome, Pfeiffer syndrome, and thanatophoric dwarfism [7]. All of these syndromes result from dominant missense mutations in FGFR genes [11]. Unlike advanced maternal age, known to be associated with chromosomal changes such as copy number variations or aneuploidy (trisomy 21 causing Down’s Syndrome) [12], advanced paternal age is associated with sequence mutations [13].
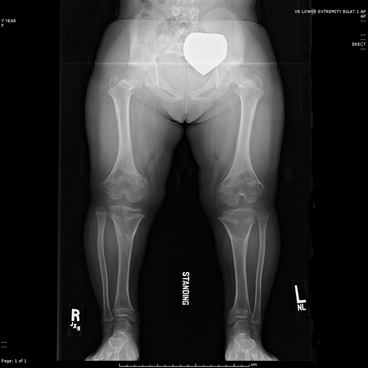
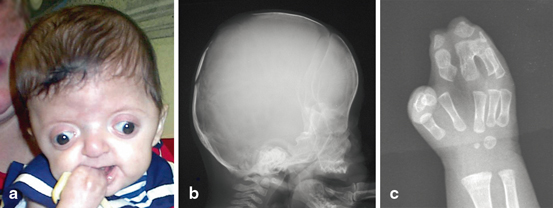
Fig. 9.2
Achondroplasia Lower limb length is 15 in (37.6 cm) in this 8 year-old child. Note the abnormal joint shape and growth plates
Fig. 9.3
Child with apert syndrome. Children with apert syndrome have abnormal development of the face (a), skull (b) and hands (c)
It is worth noting that not all sporadic disorders are associated with age effects. Krakow et al. hypothesized that mutations in the TRPV4 gene, which is known to cause autosomal dominant brachyolmia (ADB), may also cause spondylometaphyseal dysplasia Kozlowski type (SMDK) [14]; both disorders present with abnormal vertebral body development. After sequencing the gene in five sporadic patients, they identified a missense mutation (R594H) in four patients; this mutation was confirmed to be de novo in two patients. The final sporadic patient had a different de novo mutation (A716S). In another example, gene sequencing of sporadic patients with fibrodysplasia ossificans progressiva (FOP) (Fig. 9.4) identified a de novo missense mutation (R206H) in the ACVR1 gene in a young patient [15]. The ACVR1 gene was initially identified using genome-wide linkage analysis in multiple families with dominant inherited FOP [16]. In this study, the authors identified the R206H mutation inherited among all patients in seven families as well as in 32 patients with sporadic FOP.
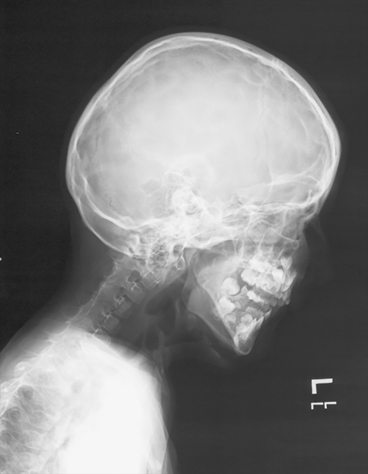
Fig. 9.4
Fibrodysplasia Ossificans Progressiva. Lateral skull and neck showing complete fusion of the posterior elements of the spine as well as characteristic soft tissue ossification in the anterior neck
Mosaic Mutations
As noted, mutations that occur after fertilization, during early embryonic development, yield a pattern of somatic mosaicism. Thus, some cells in a patient may carry the mutation while other cells do not. Therefore, the disease is not systemic, but only occurs among the “affected” cells of the patient. The extent of the disease is determined, in part, by the fraction of mutated cells expressing the mutant protein; consequently, the extent of mosaicism is expected to lead to great variability between patients. Mosaic mutations are best diagnosed using DNA from biopsies of affected tissue, and mosaic mutations are known to cause several disorders treated in orthopaedic clinics.
McCune Albright syndrome presents as a clinical association of fibrous dysplasia, precocious puberty and café-au-lait spots. It is caused by a somatic mutation in a critical enzyme that decreases GTPase activity and results in over production of cAMP. In turn, this increases proliferation of the involved cells, especially bone marrow stroma. Malignant degeneration has been reported but is not considered a high risk for this disease. This condition would be lethal if it were systemic (inherited) but is maintained as a disease because of its somatic mosaicism [17, 18].
Recently, the genetic cause of Ollier disease and Maffucci syndrome was identified. Both conditions are sporadic and present with multiple cartilaginous tumors during childhood; the skeletal severity depends on the extent and location of tumor formation (Fig. 9.5). When the enchondromatosis is associated with deep vascular lesions the condition is called Maffucci syndrome. Ollier’s and Maffucci’s diseases are known to be asymmetric in presentation and at times present with hemimelic involvement. Neither of these is transmitted genetically in a Mendelian fashion but both are known to involve dysregulation of normal enchondral ossification, which may result in malignant degeneration (Fig. 9.6). Maffucci recognized that some of his patients went on to develop cancers in the enchondromatous lesions that had been slowly growing since childhood [19]. As well, the extent of the affected bones varies between patients; ranging from single to several affected bones [20]. Based on their prior association with cartilaginous tumors, two isocitrate dehydrogenase genes (IDH1 and IDH2) were sequenced in multiple lesions from many patients [21]. Nearly all the lesions harbored mutations in one of the two genes. Additionally, identical mutations were identified among multiple tumors from the same patient, suggesting the mutations were mosaic in the patient. The somatic mosaicism of mutations in these genes explains the wide phenotypic spectrum of the disease [21].
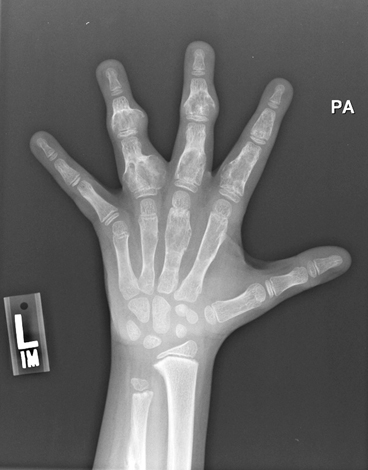
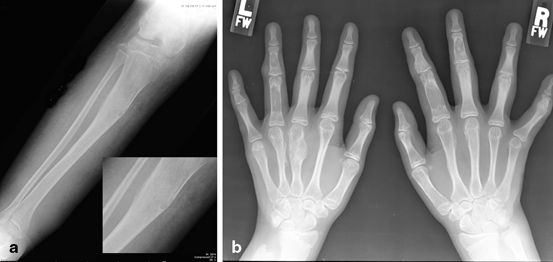
Fig. 9.5
Ollier Disease. Radiograph of the hand of a patient with Ollier disease
Fig. 9.6
Maffuci syndrome. Malignant degeneration of an enchondromatous/angioma in the a proximal tibia and b hands in an 8 year-old with Maffucci syndrome. Histology showed angiosarcoma
Similar to Ollier and Maffucci’s syndrome, Proteus syndrome is another very rare sporadic disorder. Patients present with overgrowth affecting multiple body regions and various orthopaedic abnormalities. Zhou et al. wrote in 2001 that they suspected a mutation in the PTEN pathway to explain Proteus syndrome (PTEN negatively regulates the PI3K/AKT/MTOR pathway described below) [22]. They had identified genomic mutations in this pathway for other overgrowth and hamartoma syndromes such as Cowden and Bannayan-Riley-Ruvalcaba syndrome, which are hereditary [22]. SOLAMON (segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus) syndrome, another overgrowth condition, is also known to be associated with mutations in the PTEN pathway [23]. Whole-exome sequencing (WES) was performed using DNA extracted from “affected” overgrowth lesions from multiple patients and compared to DNA from blood (considered “normal”). This comparative analysis identified a somatic mutation in the AKT1 gene that was specific to overgrowth tissue. Furthermore, AKT1 mutations were identified in multiple patients [24].
The mosaic AKT1 mutation in Proteus syndrome results in a gain-of-function effect, similar to the de novo mutations in the FGFR proteins associated with paternal age effects (described above). The Proteus mutation results in dysregulated activation of the AKT1 protein and upregulation of the PI3K/AKT/MTOR pathway. This pathway is well characterized for its involvement in various cancers and is associated with other overgrowth syndromes such as CLOVES (congenital, lipmatous overgrowth, vascular malformations, epidermal nevi and skeletal anomalies), fibroadipose hyperplasia and hemimegalencephaly [25–27].
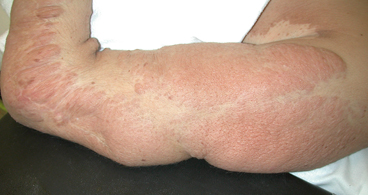
Not all mosaic disease-causing mutations result in a gain-of-function effect. Neurofibromatosis type 1 (NF1) is caused by loss-of-function mutations that disrupt the NF1 gene. Neurofibromin, the protein encoded by the NF1 gene, is a negative regulator of cell signaling; thus, loss of neurofibromin causes an increase in cell signaling. NF1 is a relatively common disorder caused either by de novo mutations in sporadic cases or inherited from affected parents. However, NF1 is also caused by mosaic mutations, which result in a distinct “segmental” NF1 phenotype. In one patient with segmental NF1, extensive freckling and café-au-lait spots were localized to one side of the patient’s body with distinct borders. Only one copy of the NF1 gene was present in a skin biopsy of one of the café-au-lait spots, but the normal two copies of the gene were present in samples from blood cells and a skin biopsy from an unaffected area of the same patient [28]. Thus, this patient was mosaic for the mutation, which manifested as a distinctly different (segmented) phenotypic presentation, compared to other NF1 patients with sporadic or inherited NF1. NF1 is also associated with both limb overgrowth and tumor formation (Fig. 9.7). Malignant degeneration is a known risk of this disease. The phenotypic variability in this disease spectrum has been attributed to the extent of DNA mutation [29]; the clinical spectrum of NF1 includes lesions in multiple organ systems. Central nervous system involvement, intracranial lesions, congenital pseudarthroses of long bones and limb overgrowth make surveillance for malignant degeneration worrisome.
 The Genetic Architecture of Idiopathic Scoliosis
The Genetic Architecture of Idiopathic Scoliosis
 Molecular Genetics of Congenital Multiple Large Joint Dislocation
Molecular Genetics of Congenital Multiple Large Joint Dislocation
 Overview of Next Generation, High-Throughput Molecular Genetic Methods
Overview of Next Generation, High-Throughput Molecular Genetic Methods
 Neurofibromin in Skeletal Development
Neurofibromin in Skeletal Development
 DMP-1 in Postnatal Bone Development
DMP-1 in Postnatal Bone Development
 Genetic and Environmental Interaction in Malformation of the Vertebral Column
Genetic and Environmental Interaction in Malformation of the Vertebral Column




Related posts:
The Genetic Architecture of Idiopathic Scoliosis
Molecular Genetics of Congenital Multiple Large Joint Dislocation
Overview of Next Generation, High-Throughput Molecular Genetic Methods
Neurofibromin in Skeletal Development
DMP-1 in Postnatal Bone Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
