Gene
OMIM number
Autosomal dominant
Larsen syndrome
FLNB
150250
Ehlers Danlos syndromes, arthrochalasia type
COL1A1, COL1A2
130060
Spondyloepimetaphyseal dysplasia with joint laxity- type 2
KIF22
603546
Autosomal recessive
Spondyloepimetaphyseal dysplasia with joint laxity type 1
B3GALT6
271640
Multiple joint dislocations—CHST3 type
CHST3
143095
Multiple joint dislocations—B3GAT3 type
B3GAT3
245600
Chondrodysplasia with joint dislocation- GPAPP type
IMPAD1
614078
Desbuquois dysplasia type 1
CANT1
251450
Desbuquois dysplasia type 2
Unknown
251450
Multiple Joint Dislocations—Reunion Island type
Unknown
245600
Autosomal Dominant Disorders
Larsen syndrome (OMIM 150250)
Larsen syndrome is characterized by a variable combination of congenital dislocations of the large joints (elbows, hips, knees, occasionally shoulders), scoliosis, cleft palate, conductive deafness and cervical kyphosis, which can be associated with cervical spinal cord damage [3]. It exhibits complete penetrance with significantly variable expressivity. The vast majority of individuals in Larsen syndrome (as defined by a phenotype shown to be caused by a mutation in FLNB) have dislocations or subluxations of the large joints (80 % hip, 80 % knee, and 65 % elbow) with subluxation of the shoulders being the mildest manifestation (Fig. 3.1a). Clubfoot is also present in 75 %. Larsen syndrome is allelic to three other disorders of defective osteogenesis—atelosteogenesis types I and III and Boomerang dysplasia –making up the so-called Larsen syndrome-atelosteogenesis “LS-AO” spectrum disorders. The latter two disorders in the spectrum are invariably lethal in the perinatal period [4]. Despite this link, stature is only mildly affected in Larsen syndrome with the height of the majority of individuals with the condition lying between the 1st and 10th centile. Radiographically evident spinal abnormalities are present in the majority with some degree of cervical spinal dysplasia noted in 50 %. Typically this takes the form of subluxation or fusion of the bodies of C2-C4, associated with posterior vertebral arch dysraphism. Individuals with Larsen syndrome and cervical spine dysplasia are at significant risk of cervical cord myelopathy and secondary tetraparesis. Other clinical findings include cleft palate and conductive deafness. Although laryngotracheomalacia has been reported in association with Larsen syndrome, few individuals with confirmed Larsen syndrome are severely affected. Survival into adulthood is the norm, and cognitive functioning is unaffected. Clinical variability can result from the presence of somatic mosaicism for a causative mutation in a mildly affected parent with the associated mutation in the germline leading to a more severe phenotype in the offspring.
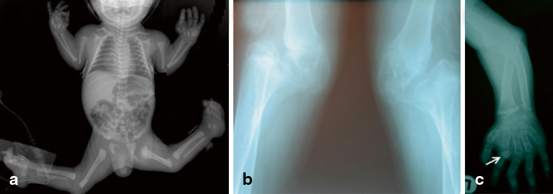
Fig. 3.1
Radiographs of individuals multiple large joint dislocation syndromes. a Larsen syndrome showing disarticulation of the knee and elbow joints, but, with the exception of distal taping of the humerus, little disturbance to the morphogenesis of the bony structures of the skeleton. b Radiographs of the knee joints of an adult individual with CHST3 deficiency show significant erosion and destruction of the joint spaces as a consequence of the epiphyseal dysplasia seen in this condition in the context of chronic joint dislocation. c Desbuquois dysplasia demonstrating shortening of the long bones, metaphyseal widening and dislocation of the radial head and advanced bone age. An accessory ossification center is evident proximal to the proximal phalanx of the second digit (arrowed)
Although the primary clinical presenting feature in Larsen syndrome is dislocation of the large joints there are no consistent primary epiphyseal or metaphyseal anomalies present. In some individuals with a clinical diagnosis of Larsen syndrome a tapering distal humerus can be observed, reminiscent of that seen in the allelic disorder atelosteogenesis III. The most characteristic radiological signs are the appearance of supernumerary carpal ossification centres evident from early childhood onwards. A bipartite calcaneal ossification centre is similarly seen but is a less specific diagnostic sign.
Larsen syndrome is caused by missense mutations or small in-frame deletions in the gene encoding the actin binding protein, filamin B [3, 4]. The level of expression of protein is unaffected by these mutations and the phenotypic dissimilarity between LS and the allelic loss of function recessive disorder, spondylocarpotarsal syndrome, has led to the conclusion that mutations leading to LS lead to disease through a gain of function mechanism. All three filamin paralogs (filamin A, B and C) encode a highly homologous tertiary structure with an N-terminal actin binding domain composed of two calponin homology domains, CH1 and CH2 followed by a chain of 24 filamin repeats, each which adopt an immunoglobulin-like barrel structure consisting of seven folded β-strands. The chain of filamin repeats is interrupted by two “hinge” regions between repeats 15 and 16 and repeats 23 and 24. Filamin B adopts a dimeric structure in vivo and in vitro, this interaction being mediated by the anti-parallel apposition of repeat 24 from each monomer.
Mutations leading to Larsen syndrome are clustered within exons encoding the actin binding domain or in the region of the gene specifying domains flanking the flexible hinge region of the filamin B protein. To date over 40 mutations have been described with some mutations being highly recurrent as de novo events in unrelated individuals (Table 3.2). For pathogenic substitutions occurring in the actin binding domain of the protein, a gain of function of mechanism has been proposed manifested biochemically by enhanced avidity for the binding partner actin [5]. A similar but more pronounced biochemical property is conferred by mutations that lead to atelosteogenesis types I and III and Boomerang dysplasia, suggesting that the defect in osteogenesis evident in these disorders might have its subclinical equivalent in individuals with Larsen syndrome. No clear explanation for how the substitutions that cluster around the hinge region confer the phenotype has been offered but these mutations have been shown to lead to a redistribution of filamin protein from a cortical to a perinuclear position [6].
Table 3.2
Mutations in FLNB leading to Larsen syndrome
Mutation | Protein | Protein domain |
|---|---|---|
c.362A > T | p.Tyr121Phe | CH2 |
c.482T > G | p.Phe161Cys | CH2 |
c.488A > C | pGln163Pro | CH2 |
c.500A > G | p.Asp167Gly | CH2 |
c.501C > A | p.Asp167Glu | CH2 |
c.508G > C | p.Ala170Pro | CH2 |
c.520C > A | p.Leu174Met. | CH2 |
c.572C > T | p.Pro191Leu | CH2 |
c.590A > T | p.Asn197Ile | CH2 |
c.622T > C | p.Trp208Arg | CH2 |
c.661A > T | p.Ile221Phe | CH2 |
c.679G > A | p.Glu227Lys | CH2 |
c.685T > C | p.Ser229Pro | CH2 |
c.700C > G | p.Leu234Val | CH2 |
c.719C > T | p.Ala240Val | CH2 |
c.4292T > G | p.Leu1431Arg | Repeat 13 |
c.4550C > A | p.Ala1517Asp | Repeat 14 |
c.4580T > A | p.Leu1527His | Repeat 14 |
c.4580T > C | p.Leu1527Pro | Repeat 14 |
c.4621G > C | p.Ala1541Pro | Repeat 14 |
c.4625T > C | p.Ile1542Thr | Repeat 14 |
c.4711_4713del | p.1571delAsn | Repeat 14 |
c.4725_4736del12 | p.1576_1579del | Repeat 14 |
c.4756G > A | p.Gly1586Arg | Repeat 14 |
c.4769T > C | p.Ile1590Thr | Repeat 14 |
c.4775T > A | p.Val1592Asp | Repeat 14 |
c.4781A > C | p.Tyr1594Ser | Repeat 14 |
c.4795A > T | p.Ile1599Phe | Repeat 14 |
c.4805C > A | p.Ser1602Tyr | Repeat 14 |
c.4808C > T | p.Pro1603Leu | Repeat 14 |
c.4936G > A | p.Gly1646Ser | Repeat 15 |
c.5023_5025delTTC | p.1675delPhe | Repeat 15 |
c.5047A > T | p.Thr1683Ser | Repeat 15 |
c.5071G > T | p.Gly1691Cys | Repeat 15 |
c.5071G > A | p.Gly1691Ser | Repeat 15 |
c.5072G > A | p.Gly1691Asp | Repeat 15 |
c.5095C > G | p.Pro1699Ala | Repeat 15 |
c.5706C > A | p.Ser1902Arg | Repeat 17 |
c.5705C > A | p.Ser1902Ile | Repeat 17 |
LS and its allelic disorders, atelosteogenesis types 1 and 3 and Boomerang dysplasia, share significant phenotypic similarities to the filaminopathies cause by mutations in the paralogous gene FLNA, although notably joint dislocations are not a feature of this group of disorders [7]. Although the differing expression of FLNB and FLNA over the growth plate may explain some of the differences in the chondrodysplasia seen in the FLNA-linked otopalatodigital syndrome spectrum disorders and the LS-AO spectrum [4], the role of FLNB in the formation and integrity of the joint capsule, possibly underlying the multiple congenital joint dislocations observed in these conditions, has not been investigated. Within the growth plate of individuals with AO1 there has been the frequent observation of multinucleate giant cells in the context of a relatively cell sparse structure. This suggests that FLNB exerts some role in cell proliferation or division, and indeed FLNB has been observed lining the cell cleavage furrow of dividing chondrocytes in normal human growth plate [4].
The Ehlers Danlos Syndrome (EDS) Arthrochalasia Type
The group of conditions broadly subsumed under the term Ehlers Danlos syndromes phenotypically manifest with connective tissue dysfunction leading to fragile, thin or wrinkled skin, joint hypermobility and occasionally fragility of other organ systems such as the vasculature. In the Villefranche classification of EDS [8], six main descriptive types were substituted for earlier types numbered with Roman numerals. One form in particular, the arthrochalasia type (formally termed EDS VII) is characterised by hip dislocation. This disorder is caused by a genetically determined resistance to enzymatic procollagen maturation. Milder forms of EDS (EDS hypermobility type) and familial joint instability syndrome are more associated with pronounced joint laxity and congenital joint dislocation is uncommon .
EDS, arthrochalasia type, is distinguished clinically from the other types of EDS by the frequency of congenital hip dislocation, extreme joint laxity with recurrent joint subluxations but minimal skin involvement [9, 10]. Dislocation of the knees and other large joints can occasionally be observed [11], the fingers can exhibit swan neck deformities and contractures and inguinal herniae have recurrently been observed. Radiographic manifestations can include calvarial wormian bones and recurrent fractures. EDS, arthrochalasia type, is caused by either monoallelic mutations in COL1A1 and COL1A2 [12–15] where they lead to the exclusion of exon 6 which encodes procollagen N-proteinase cleavage site of each respective gene transcript [9]. Interestingly, biallelic mutations in the gene encoding the procollagen protease convertase itself, ADAMTS2, cause EDS dermatospraxis type, which is characterised by a severe skin phenotype and joint hypermobility, but only rarely congenital joint dislocation. The presumptive pathogenesis of this disorder presumably relates to the role that collagen 1 plays in the integrity of the articular joint capsule of the large synovial joints .
Spondyloepimetaphyseal Dysplasia with Joint Laxity-2
The principal clinical characteristics of SEMDJL2 (previously termed SEMD-JL- leptodactylic type or SEMD-JL-Hall type) include generalized ligamentous laxity (especially evident at the knees), short stature, maxillary retrusion and mild spinal deformities [16]. Dislocations can occur at all the large joints but the knees are the most commonly and severely affected joint. Laryngotracheomalacia in infancy and childhood has been reported in a sizable minority of cases [17, 18] and occasionally necessitating tracheostomy placement [19]. Marked intrafamilial phenotypic variability has been observed. Features that are noticeably absent in described cases to date that might aid in differential diagnosis, particularly from Larsen syndrome , include cleft palate, deafness and congenital kyphoscoliosis. Key radiographic findings include small, flattened, irregular and fragmented epiphyses and widened, irregular metaphyses with striations. The dorsal vertebral bodies show posterior decrease in height, vertebral endplate irregularity, and progressive caudal narrowing of the interpedicular distances. The metacarpals are gracile with long slender phalanges and the distal phalangeal tufts are prominent. The carpal bones are small and irregular and progressively erode over adolescence and into adulthood. There is a global epiphyseal delay in ossification, especially evident in the phalanges [20]. The disorder evolves into an epiphyseal dysplasia with the precocious development of osteoarthritis.
SEMD-JL2 is caused by heterozygosity for mutations in KIF22 which encodes KIF22, a kinesin motor domain-containing protein that has homology to proteins that mediate intracellular transport along microtubular structures [20, 21]. Mutations are exclusively missense, are recurrent and all mutations reported to date occur in exon 4 which encodes highly conserved residues which are adjacent to an ATP binding site within the motor domain of the protein. Notably KIF22 binds to chromosomes and appears to have functions relating to chromosomal segregation during certain phases of mitosis [22, 23]. It is perhaps notable that a cell cycle related function is implicated in the pathogenesis of a joint dislocation disorder, just as some observations relating to the pathogenesis of some of the LS-AO spectrum syndromes also indicate a disturbance in cell division.
Autosomal Recessive Disorders
Spondyloepimetaphyseal Dysplasia with Joint Laxity Type 1
Originally described in South African families of Afrikaans descent [24], Spondyloepimetaphyseal dysplasia with joint laxity type 1 (SEMDJL1; previously termed SEMDJL-Beighton type) is characterized by the cardinal skeletal findings of vertebral abnormalities and ligamentous laxity. This disorder evolves a progressive severe kyphoscoliosis, often resulting in severe respiratory compromise. Large joint manifestations include dislocation of the radial head and dislocated hips. Clubfeet are commonly observed and a subset of patients can also present with bone fragility [25]. Joint hypermobility is particularly evident in the hands and a dysmorphic facial profile has been proposed (an oval face, with maxillary flattening, prominent eyes and a long philtrum). Cleft palate is commonly observed [24, 26–28]. Accompanying features included skin fragility, delayed wound healing, joint hyperlaxity and contractures and intellectual disability. The progressive kyphoscoliosis associated with this disorder is especially refractory to external bracing and internal fixation is often indicated. Progression can often not only result in cardiorespiratory compromise but also spinal cord compression. Death within the first two decades of life is common although survival into the fourth decade has been reported [29]. The radiological phenotype of this condition consists of the severe scoliosis , ‘bat-like’ splaying of the ilia, dysplastic acetabulae and brachydactyly. In some individuals expanded metaphyses can be present [30, 31].
Related posts:
 Insights into the Genetics of Clubfoot
The Genetic Architecture of Idiopathic Scoliosis
Overview of Next Generation, High-Throughput Molecular Genetic Methods
Neurofibromin in Skeletal Development
DMP-1 in Postnatal Bone Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column
Insights into the Genetics of Clubfoot
The Genetic Architecture of Idiopathic Scoliosis
Overview of Next Generation, High-Throughput Molecular Genetic Methods
Neurofibromin in Skeletal Development
DMP-1 in Postnatal Bone Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


