Fig. 8.1
Radiographs showing (a) a case of SCDO with severe vertebral segmentation anomalies throughout the vertebral column (adapted from [51] with permission from Oxford University Press); and (b) a case of congenital scoliosis with a few vertebral and rib anomalies and marked scoliosis. Arrows indicate malformed vertebrae and arrowheads indicate missing ribs. (Adapted from [54] with permission from Elsevier)
The Developmental Origins of Vertebral Defects
The axial skeleton develops from precursor tissues called somites. These are blocks of mesodermal tissue that develop along the axis of the embryo in a regular repeated process of segmentation from an unsegmented precursor tissue, the presomitic mesoderm (PSM, summarized in Fig. 8.2). Vertebral defects can arise either from a disruption of the segmentation process, or from a failure of the correct differentiation of the somite. Somite formation or somitogenesis occurs via a complex interplay of FGF, WNT, Notch and retinoic acid signal transduction pathways [42]. Failure of this process in both humans and mice can result in a variety of phenotypes including vertebral fusion, hemi-vertebrae, and proximal fusions or bifurcations of the ribs. These can be considered as “dysostoses”. Mature somites consist of two cellular compartments [11]. Firstly, cells derived from the sclerotome form the vertebrae and ribs, with a subgroup of cells within this domain going on to form the tendons (syndetome). Secondly, the dermomyotome consists of two populations of cells: cells from the dermotome form the dermis of the trunk, and cells from the myotome form the axial musculature. Rib and vertebral defects can also arise from a failure of sclerotome differentiation. However the phenotypes generated by such disruption are subtly different to those generated by segmentation defects. For example, the vertebral spinal processes fail to fuse medially, instead fusing in a rostral-caudal direction, and rib fusions and bifurcations generally occur distally [32, 48, 67]. These can be considered as “dysplasias”.
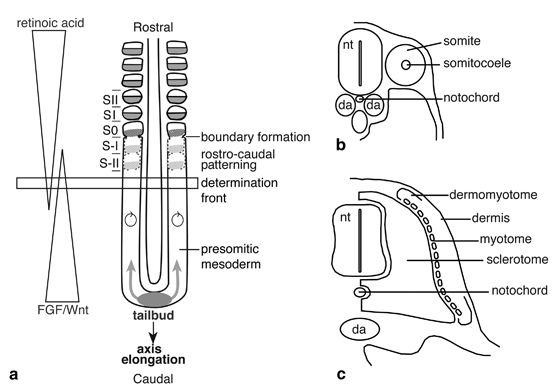
Fig. 8.2
Somitogenesis in mouse. a Schematic representation of somite formation and rostral-caudal patterning in mouse. A dorsal view of a midgestation mouse embryo showing the production of presomitic mesoderm from the tail bud, resulting in elongation of the embryonic axis. FGF and Wnt signaling gradients emanate from the tail bud, while a retinoic acid signaling gradient derives from the somites. These opposing gradients set the position of the determination front: cells rostral to this front become competent to form somites. Notch1 signaling oscillates (circle with arrow) between on and off states in PSM cells, giving the impression that a wave of activity sweeps rostrally. This wave stalls in the region of the next forming somite, forming a sharp band of cells undergoing signaling (S-I). Rostral-caudal polarity is established in presumptive somites (S-II, S-I) and a boundary forms (between S0 and S-I), defining the formation of a nascent epithelial somite. Rostral-caudal polarity is maintained as somites mature and differentiate along their dorsoventral axis to form dermomyotome and sclerotome. b Cross-sectional view of a newly formed epithelial somite. c Cross-sectional view of a mature somite after dorsoventral compartmentalization and differentiation. The dermomyotome has largely differentiated into the dermis and the myotome. The sclerotome has expanded and moved ventrally to surround the neural tube and notochord. SI and SII, segmented somites; S0, next somite to form; S-I and S-II, prospective somites; nt neural tube; da dorsal aorta. (Figure and legend reproduced from [54] with kind permission from Springer Science and Business Media)
Autosomal Recessively Inherited Severe Non-Syndromic Multiple Contiguous Vertebral Defects
Over the past 13 years it has become apparent that congenital vertebral defects can arise from simple genetic deficiencies, from maternal or embryonic exposure to environmental teratogens, or a combination of both factors. These etiological insights have come from a combination of human molecular genetics and animal studies. The most significant advances in identifying the genetic etiologies of human congenital vertebral malformations have resulted from studying familial cases with clear autosomal recessive inheritance patterns (Table 8.1). This is in a large part due to genetic mapping and candidate gene sequencing of consanguineous cases. In such cases, the causative mutation has been inherited by both parents from a recent common ancestor, and is therefore relatively easy to identify. In addition, many of details of the underlying genetic pathways and molecular mechanisms of vertebral formation have now been uncovered using a variety of animal model systems. These studies have identified a large number of candidate genes for causing vertebral defects (reviewed in [53]), and this has accelerated the discovery of the genetic bases of many human cases of vertebral defects. One particular disorder, spondylocostal dysostosis (SCDO) , has been intensely investigated. Cases of SCDO have abnormal segmentation of nearly all vertebrae with associated abnormal ribs. However, even though the vertebral defects are severe, the rightward and leftward curves tend to cancel each other out resulting in a mild degree of non-progressive scoliosis without a major asymmetry of the thorax. The phenotype can result in compromised respiratory function in neonates due to the reduced capacity of the thorax, respiratory complications in children, and an increased incidence of inguinal hernia in males. However, despite the dramatic vertebral phenotype, SCDO cases rarely have any neurological affects. Below we outline the genetically-defined sub-classes of SCDO.
Table 8.1
Summary of human vertebral malformation syndromes with known etiology
Disease name | Inheritance | OMIM | Gene | References |
|---|---|---|---|---|
SCDO1 | AR | 277300 | DLL3 | |
SCDO2/STD | AR | 608681 | MESP2 | |
SCDO3 | AR | 609813 | LFNG | [50] |
SCDO4 | AR | 613686 | HES7 | |
SCDO5 | AD | 122600 | TBX6 | [56] |
KFS2 | AR | 214300 | MEOX1 | [38] |
KFS1 | AD | 118100 | GDF6 | |
KFS3 | AD | 613702 | GDF3 | [68] |
Congenital scoliosis | AD | Na | MESP2 HES7 | [54] |
SCDO1 (OMIM 277300)
Although severe non-syndromic multiple contiguous vertebral defects had been described in the literature as early as 1938 [30], it was not until the end of the twentieth century that the first insights into their genetic etiology were made, arising from the combined efforts of three groups. Turnpenny and colleagues approached the problem by using the then relatively new method of homozygosity mapping [34] on three large consanguineous families to map the SCDO critical region in these families to a 7.8 cM region of chromosome 19 [7, 59]. Simultaneously, Kusumi and colleagues were investigating a recessive mouse mutant pudgy that had come from an X-ray mutagenesis screen at the Oak Ridge National Laboratory prior to 1961. This mouse line had a rib and vertebral phenotype very similar to that seen in human cases of SCDO, and a combination of classical genetic mapping and BAC transgenic rescue methods narrowed the pudgy critical region to a 159 kb region containing eight genes [33]. One of these was Dll3 encoding the delta-like 3 protein, a gene that had been shown by Dunwoodie and colleagues the previous year to be expressed in the vertebral precursor tissues [18]. Sequence analysis of Dll3 in the mutant mice revealed a frameshift mutation that was not present in the parental mouse strain. Kusumi and Turnpenny then collaborated to demonstrate that in humans DLL3 lay in the SCDO critical region on chromosome 19, and that each of the three families used for homozygosity mapping had different deleterious mutations in DLL3 [7]. This discovery was quickly translated to clinical practice, and routine sequencing of DLL3 in SCDO cases by clinical services is now standard in many countries internationally. Around 70 % of SCDO cases have homozygous mutations in DLL3, and up to 40 distinct mutations have been identified [62], making this the most important gene causing SCDO.
SCDO2 (OMIM 608681)
Investigation of a small consanguineous family, also with an autosomal recessive inheritance pattern, resulted in the discovery of the second SCDO-associated gene [66]. The proband had extensive vertebral defects throughout the spine, however the phenotype was distinct from that of SCDO1 in that the ribs were well aligned. Homozygosity mapping of two affected siblings, their parents and an unaffected sibling detected a 36.6 Mb region on chromosome 15. This region contained more than 50 genes, including an excellent candidate MESP2 encoding mesoderm posterior 2 homolog protein. Mouse embryos homozygous for a targeted null allele of Mesp2 show very similar vertebral and rib malformations to homozygous pudgy mice [44]. Sequence analysis of MESP2 in the family detected a 4 bp insertion that segregated with disease. This mutation causes a frameshift and premature truncation of the MESP2 protein, however it is not predicted to cause nonsense-mediated decay of the transcript. MESP2 is a transcription factor required for formation of the vertebral precursors (somites). When tested in an in vitro transcription activation assay, the mutant MESP2 protein was significantly less active than the wildtype protein [55]. Taken together, these data provide good evidence that mutation of MESP2 is causative of the vertebral phenotype in this family.
Interestingly, mutation of MESP2 causes another human congenital vertebral segmentation abnormality, spondylothoracic dysostosis (STD) , that is phenotypically distinct from SCDO [4]. Typically in STD, the thoracic spine and the ribs are most affected, with the latter being fused posteriorly creating a fan-like or “crab” configuration. This results in a severe reduction of respiratory function, and 75 % of cases die in infancy. In addition, many cases have fusion of all cervical vertebrae, as well as that of C1 to the skull, preventing the head from being turned. STD is particularly prevalent in Puerto Rico, most likely due to a founder effect. Cornier and colleagues [14] used a candidate gene sequencing approach to determine the genetic cause of STD. They sequenced five genes (DLL3; LFNG; MESP2; HES7; and PSEN1) in a large cohort of patients with congenital vertebral defects. In patients with STD they identified three mutations in MESP2 predicted to either produce non-functional proteins and/or to be susceptible to nonsense-mediated RNA decay (NMD). An in vitro transcription assay was used to confirm the deleterious nature of these mutations.
Despite being caused by mutation of the same gene, SCDO2 and STD clearly have different phenotypes. This is especially apparent in the ribcage. It has been suggested that these phenotypic differences might be the result of different types of MESP2 mutation (P. Turnpenny, personal communication). It is also apparent that there are clear phenotypic differences between SCDO1 and SCDO2 in humans. Likewise, comparisons of Mesp2 and Dll3 homozygous null mouse embryos show differences at the molecular level in somite patterning. Mesp2 null embryos lack specification of the rostral half of the mature somite [44], whereas Dll3 null embryos express markers of both rostral and caudal somite identity, but lack the correct spatial separation of the two [19]. The rostal and caudal compartment of the mature somite give rise to different parts of the mature vertebra, this may explain the phenotypic differences seen in human cases of SCDO1 and SCDO2.
SCDO3 (OMIM 609813)
A third SCDO causative gene was identified in a consanguineous case of SCDO with a severely disorganized spine including malformations throughout the cervical, thoracic and lumbar spine [50]. There were no mutations detected in DLL3 or MESP2, and so a candidate gene sequencing approach was used to identify the causative mutation. LFNG, encoding the LFNG O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase protein, was chosen for sequencing because mouse embryos carrying homozygous null mutations of Lfng have a similar vertebral and rib phenotype to that of Dll3 and Mesp2 homozygous null mutants [20, 69]. A homozygous single nucleotide change predicted to cause a non-synonymous F188L amino acid change was present in the affected individual, and both parents were heterozygous carriers for the variant. The functional consequences of this sequence alteration were tested in a series of in vitro assays, showing that the variant was likely to be highly deleterious to protein function. To our knowledge only one other mutation in LFNG has been discovered (Bonafe, unpublished data), and therefore mutation of LFNG is likely to be the least common cause of SCDO.
SCDO4 (OMIM 613686)
Advances in molecular genetic techniques have greatly facilitated homozygosity mapping of disease loci in offspring of consanguineous unions. First SNP arrays, and now whole-exome sequencing, have allowed much more rapid identification of new loci causing genetic diseases. We investigated a single affected individual with consanguineous parents who had multiple and contiguous vertebral segmentation defects mainly involving the thoracic spine and ribs, but also affecting the cervical and lumbar regions. The patient also had Chiari II malformation, lumbo-sacral myelomeningocele, ectopic stenotic anus and talipes. We used SNP array technology to map a single large “homozygous-by-descent” region unique to the affected individual [51]. This region of 10.1 Mb contained 201 genes, of which two (HES7 and DVL2) were possible candidates on the basis of the skeletal phenotype of homozygous null mouse embryos for these genes [5]. Sequence analysis revealed a homozygous non-synonymous change in the proband at a highly evolutionarily conserved residue in the DNA-binding domain of HES7 encoding hairy and enhancer of split 7. The functional consequences of this nucleotide variation were tested in two in vitro assays, showing that this significantly reduced the transcriptional repression activity of the protein.
Three more deleterious mutations in HES7 have now been reported [51, 52, 55]. Closer analysis of the phenotypes of all ten reported cases suggests that there are two additional phenotypes associated with homozygous deleterious mutations in HES7 that have not been reported in any SCDO cases involving mutation of DLL3, MESP2 or LFNG [56]. Firstly, three of the ten cases had neural tube defects including myelomeningocele, spina bifida occulta and/or Chiari type II malformation. Secondly, four of the ten cases had dextrocardia with situs inversus. Neural tube malformations have been reported in patients with SCDO with no reported molecular diagnosis [8, 13, 23, 46] and three of these patients were also reported as having laterality defects [8, 13, 23, 46]. Thus is it possible that these are also cases involving HES7 mutation.
Autosomal Dominantly Inherited Severe Non-Syndromic Multiple Contiguous Vertebral Defects
Autosomal dominantly inherited SCDO cases are much more rare than the autosomal recessive forms, possibly representing less than 10 % of total cases [39]. To date the genetic lesion for an autosomal dominant form of SCDO has only been identified in a single family [56] (Table 8.1).
SCDO5 (OMIM 122600)
We investigated a family with affected individuals in three consecutive generations. All individuals had multiple generalized contiguous vertebral malformations throughout the spine, including hemivertebra and fused vertebral blocks, however the ribs were relatively unaffected [24]. Sequence analysis of DLL3, MESP2, LFNG and HES7 was negative. We used whole-exome sequencing of three affected and two unaffected individuals to identify the causative mutation in the cases [56]. After filtering, six potentially disease-causative variants remained, one of which was in TBX6. Studies in mouse have shown that this transcription factor is an important element of the somitogenesis machinery, and mouse embryos homozygous for null or hypomorphic alleles of Tbx6 have SCDO-like phenotypes [9, 64, 65]. The mutation was confirmed by Sanger sequencing to segregate with the vertebral phenotype, and an in vitro functional assay revealed a statistically significant impairment of the transcriptional activation activity of Tbx6 protein.
Although it appears clear that TBX6 is haploinsufficient in this family, and results in a severe vertebral phenotype, it is puzzling that Tbx6 haploinsufficiency does not produce a SCDO-like phenotype in mouse. This may be due to unknown genetic modifier genes, but can only be resolved when more human cases of TBX6 mutation are reported.
Multiple Regional Vertebral Defects
The next most severe class of vertebral defects comprises those cases restricted to one or more regions of the spine. The most common of these is Klippel-Feil syndrome (KFS) , which is a general term used to refer to vertebral defects that are mostly limited to the cervical region [58]. Cases present with a short neck with limited movement due to fusions of the neck vertebrae, which can vary considerably in their number and location. Additionally, other skeletal defects are sometimes associated with KFS including Sprengel’s deformity (one scapula higher than the other) and presence of omo-vertebral bones (a bony connection from the scapula to a cervical vertebra). A significant proportion of KFS cases have two other phenotypes: deafness and a low posterior hairline. In familial cases, both autosomal dominant and autosomal recessive inheritance patterns have been described (Table 8.1).
Autosomal Recessive KFS2 (OMIM 214300)
A large consanguineous family with five affected individuals was investigated. The index case had a short immobile neck with C2-C3 fusion, Sprengel’s deformity and a low posterior hairline. In this family a single 16.1 Mb region containing 477 genes was identified by homozygosity mapping following SNP array genotyping , and was confirmed by classical linkage analysis [38]. Candidate gene sequencing revealed a single base deletion in the MEOX1 gene that segregated completely with disease. This deletion was predicted to cause a frameshift and introduce a premature termination codon. MEOX1 was also sequenced in an unrelated consanguineous family with two affected individuals. Both cases had a short neck with severely limited mobility due to cervical vertebral fusion, a low posterior hairline, scoliosis due to deformity of the thoracic spine between T2 and T11 and the presence of omo-vertebral bones. In addition they had craniofacial defects including micrognathia, cleft palate (in one case only) and congenital ptosis. A different single base change in MEOX1 was detected that was also predicted to cause a premature termination codon. Both premature stop codons were demonstrated to trigger NMD in affected individuals. MEOX1 is an excellent candidate for being the causative gene in these cases, because targeted deletion of Meox1 causes similar skeletal phenotypes in mouse [48]. Mouse embryos lacking Meox1 have entirely remodeled cranio-cervical joints such that the skull rests on the axis, rather than the atlas, and the spine is often partially fused to the skull. In addition they have variably penetrant vertebral defects, particularly in the lumbar and sacral regions, as well as occasional rib defects. However, unlike the other mouse models of vertebral malformation mentioned above, mutation of Meox1 appears to have no effect on somitogenesis . Expression patterns and levels of segmentation genes such as Mesp2, Lfng, Dll1, Dll3 and EphA4 are normal in the presomitic mesoderm [36, 48]. Instead these mutants have reduced cellular proliferation in the caudal somitic compartment of the most rostral somites. Therefore the vertebral and rib fusions are likely to be a result of impaired sclerotomal formation and differentiation. This may be a consequence of downregulation of the Meox1 direct target genes Tbx18, Uncx and Bapx1 [48]. Interestingly, the reduction in somitic cell proliferation is reduced as development proceeds, and by E12.5 proliferation has returned to normal. This may explain why the cervical vertebrae are the most affected in Meox1 mutants in both mice and humans. In conclusion, it would appear that, in contrast to SCDO/STD, KFS is a dysplasia with ongoing abnormalities of bone and/or cartilage formation during pre- and postnatal life.
Autosomal Dominant KFS1 (OMIM 118100)
The first insights into the etiology of autosomal dominant KFS came from studies of a large four-generation family. All affected individuals had C2-3 vertebral fusions, with a variable rate of more caudal vertebral fusions. In addition, affected individuals had restricted limb joint flexibility, moderate hearing impairment and severe vocal impairment. Cytogenetic analysis revealed an inversion on the long arm of chromosome 8 that segregated completely with affected status [12]. The breakpoints were either side of the GDF6 locus, and the proximal breakpoint was suggested as lying in a GDF6 regulatory region [40, 57].
Growth/differentiation factor 6 (GDF6) is a member of bone morphogenetic protein (BMP) family sub-class of the transforming growth factor β (TGF-β) super-family of secreted growth factors that have important roles in embryonic development. Candidate gene sequencing of GDF6 in an unrelated family and two “sporadic” cases of KFS revealed heterozygous non-synonymous coding variants in GDF6. In a second study, GDF6 was selected as a candidate gene for causing eye and/or vertebral defects on the basis of human linkage mapping of human syndromes including either vertebral or eye phenotypes [3]. GDF6 was sequenced in a very large cohort of cases, and seven potentially deleterious heterozygous base changes identified. Four of these were associated with solely ocular phenotypes, two with skeletal phenotypes, and the last with either phenotype. Lastly a third study found more heterozygous non-synonymous coding variants in human cases of mostly ocular, but also vertebral defects [16].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


