Fig. 2.1
Bone development cartoon (reprinted from publication title: Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends in Genetics Volume 19, Issue 8, August 2003, Page 459, with permission from Elsevier) Intramembranous ossification and endochondral ossification. a–d Craniofacial bones are formed directly from condensations of mesenchymal cells without the formation of a cartilage intermediate, a process described as intramembranous ossification. a In mice, formation of frontal bones starts at embryonic day 12.5 (E12.5) in mesenchymal condensations on the lateral side of the head. b From the mesenchymal condensation, the cellular mass with osteogenic activity spreads upward toward the top of the skull (arrows). Osteoblasts differentiate from mesenchymal condensation and produce bone matrix at E14.5. c At E15.5, the ends of the cellular masses that originate on each side meet at the midline where a suture is formed. d In the suture, cells in the mesenchymal condensations differentiate into osteoprogenitor cells. Osteoprogenitor cells then differentiate into osteoblasts that produce bone-matrix molecules. Osteoblasts differentiate into osteocytes, which become embedded within the bone matrix. e A schematic frontal view of a mouse skull (E18.5) stained with alizarin red and alcian blue. Asterisks indicate osteogenic fronts of frontal bones. f–j By contrast, long bones are formed by endochondral ossification. f Development of endochondral bones starts with the formation of mesenchymal condensations. g Cells in the mesenchymal condensations then give rise to chondrocytes whereas cells at the periphery of the condensations form a perichondrium. h Centrally localized chondrocytes proliferate actively before exiting the cell cycle and differentiating into hypertrophic chondrocytes. The sequential differentiation of chondrocytes establishes a unique cellular organization that constitutes the epiphyseal growth plate. At the distal end of the epiphyseal growth plate, hypertrophic chondrocytes further mineralize their own cartilaginous matrix. In parallel to this multistep differentiation pathway, cells from a thin layer of mesenchymal cells, called the periosteum, that envelops the cartilage matrix and invades the zone of hypertrophic chondrocytes in addition to blood vessels and osteoclasts. Whereas osteoclasts degrade the matrix of the zone of hypertrophic chondrocytes, osteoblasts deposit a bone-specific matrix. (i) The partially degraded, mineralized cartilage matrix forms a template for deposition of bone matrix by osteoblasts. Endochondral bone contains an outer envelop of cortical bone (the bone collar) that surrounds the marrow cavity and growth plate. The cortical bone is contiguous with the perichondrium. j A mouse humerus stained with alizarin red and alcian blue
Bone Complications in NF1
Short Growth and Generalized Low Bone Mineral Density
The most frequent types of skeletal manifestations in NF1 are short stature and generalized osteopenia or osteoporosis. In adults with NF1, 20–30 % exhibit height below the 3rd centile [7, 8]. How NF1 haploinsufficiency causes short stature is currently unknown. It is interesting that Nf1 heterozygous mice do not show shortening of skeletal elements or reduced bone volume. However, shortening of skeletal elements was observed in conditional mouse mutants that are deficient for Nf1 in the central nervous system (CNS), including the hypothalamus [9]. In these mice, reduction in pituitary growth hormone synthesis presumably caused a decrease in growth. Reduced growth was also observed in mice in which Nf1 was doubly inactivated in cartilage [10, 11]. Thus, short stature in NF1 may be related to deficiency in both endocrine and bone growth plate systems. Short stature itself likely does not cause significant morbidity in NF1, and growth remains proportional. However, functional and societal impact of short stature in NF1 has not been adequately investigated.
Decreased bone mineral density (BMD) has been observed in as much as 50 % of individuals with NF1 [12–19]. Generalized low bone mass and osteoporosis may be more problematic in these patients, potentially increasing risk of fractures. While literature suggests that children with NF1 have generally low BMD, the sizes of the studied patient groups are small and the clinical consequences are not well known [19]. In one study of 460 NF1 individuals there was a 5.2-fold increased risk of fractures in individuals with NF1 above 41 years of age, a 3.4-fold increased fracture risk in NF1 children, and no increased fracture risk in the age group 17–40 years [20]. In another study of 256 children with NF1 ages 5–20 years, the NF1 individuals without a focal long bone dysplasia did not have a difference in the prevalence of ever having a fracture compared to healthy controls [21]. Thus, it appears that individuals with NF1 have an age-dependent increase of fracture risk. It appears also that individuals with NF1 have generally low vitamin-D concentrations, which could be one of the factors leading to decreased BMD [22, 23]. A small study of bone biopsies from 14 NF1 individuals showed decreased trabecular bone volume and osteoidosis, which coincided with increased osteoblasts and osteoclast number [24]. These observations are similar to the findings in Nf1Col1−/− mice in which Nf1 was inactivated in the osteoblast lineage [25]. Inactivation of Nf1 in osteoblasts causes increased collagen synthesis but inhibits bone mineralization, resulting in the profound osteoidosis. Nf1Col1−/− mice show osteoblast-mediated increase of osteoclast number. More precisely the Nf1 −/− osteoblasts overproduce RANKL, the receptor activator for nuclear factor kappa-B ligand that triggers osteoclastogenesis. It has been shown that in the Nf1 deficient osteoblasts, activation of Ras triggers downstream activators ERK1/2, RSK2, PKA as well as ATF4, accelerating collagen synthesis and causing overproduction of RANKL [25]. Defective osteoblast differentiation therefore appears to be one of the main causes of bone pathology in NF1.
FGF Signaling and c-Type Natriuretic Peptide
Recent research has established a link between short growth in Nf1Col2−/− mice and increased signaling mediated by the fibroblast growth factor receptor (Fgfr) [26]. These mice lack neurofibromin (Nf1) in type II collagen-expressing cells and show a number of phenotypes reminiscent of the ones observed in mice characterized by FGFR gain-of-function mutations. In chondrocyte cultures in-vitro, Fgfr activation by FGF2 pulse-stimulation triggered rapid ERK1/2 phosphorylation in wild type (wt) and Nf1-/- chondrocytes. However, return to the basal level of ERK activation was delayed in Nf1-/- cells, suggesting that prolonged ERK signaling is one factor in deficient chondrocyte growth. In separate experiments, Nf1 Col2−/− mice were treated with the NC-2 compound, which is a derivative of c-natriuretic peptide (CNP) [26]. CNP is known to bind to the natriuretic peptide receptor B (N-PRB) and stimulate cGMP synthesis, thus suppressing MAPK activation (Fig. 2.2) [27]. The subcutaneous injections of NC−2 compound at 300 mg/kg per day throughout 18 days from birth on resulted in almost complete rescue of the growth defect in Nf1Col2 mice, suggesting CNP is a valid candidate compound for the treatment of growth deficiency in NF1. Further studies of CNP in other NF1 mouse models will be helpful to access its potential side effects and determine its suitability for the treatment of other bone manifestations.
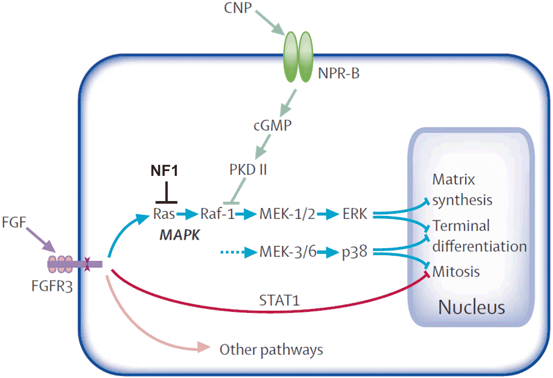
Fig. 2.2
CNP—MAPK pathway (reprinted with modification from publication tite: Achondroplasia, The Lancet Vol. 370 July 14, 2007 page 163, with permission from Elsevier). Signalling pathways of FGFR3 most relevant to growth plate chondrocytes FGFR3 signals propagated through STAT1, MAPK-ERK, MAPK-p38, and other pathways inhibit chondrocyte proliferation, post-mitotic matrix synthesis, and terminal (hypertrophic) differentiation. The CNP-NPR-B pathway and NF1 inhibit the MAPK pathways. (Reprinted from The Lancet, 370, Horton WA, Hall JG, Hecht JT, Achondroplasia, pp. 162–172, 2007, with permission from Elsevier)
Long Bone Dysplasia
Apart from generalized bone disease, about 3–5 % of individuals with NF1 develop a focal skeletal dysplasia of the long bones, predominantly the tibia (Fig. 2.3) [28]. It is currently unknown why the tibia is most often affected. The anterolateral bowing typically is identified in infancy, or even prenatally, and is usually unilateral [28]. This suggests that long bone dysplasia has a prenatal origin. Radiographically, tibial bowing in NF1 prior to fracture usually appears as cortical thickening and medullary canal narrowing at the apex of the convexity, typically near the junction of the middle and distal thirds of the tibia (Fig. 2.3b) [29]. The bowed long bone frequently sustains a fracture that is recalcitrant to union resulting in pseudarthrosis (Fig. 2.3c). Mouse experiments suggest that dystrophic bone changes are caused by complete NF1 loss-of-function in dividing and multipotent bone cells. Supporting this hypothesis, somatic (i.e. genetic alteration occurring after conception) homozygous inactivation of NF1 was demonstrated in the pseudarthrosis tissue of one NF1 patient with tibial pseudarthrosis [30]. A mouse model that recapitulates most features of NF1 tibial dysplasia is the Nf1Prx1−/− mouse (Fig. 2.4), in which Nf1 was inactivated in the limb bud mesenchyme and head mesenchyme [10]. In these mice, all cells of mesenchymal origin carry biallelic Nf1 inactivation, including bone cells, cartilage , muscle , endothelium, fat and connective tissue cells. Although the Nf1Prx1 −/− mouse was generated initially as a model of long bone dysplasia, this model has subsequently provided additional insights that relate to the broad spectrum of NF1 musculoskeletal findings. Some of the important observations from this and other mouse models pertaining to developmental processes controlled by Nf1 are described below; see also Table 2.1 for review of mouse models and bone fracture related observations.
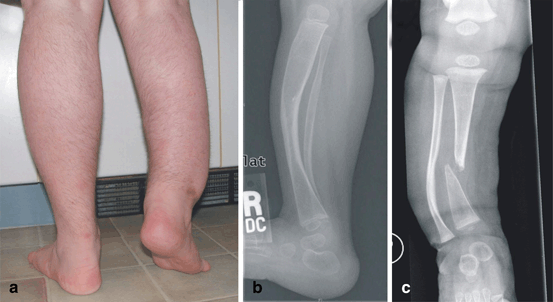
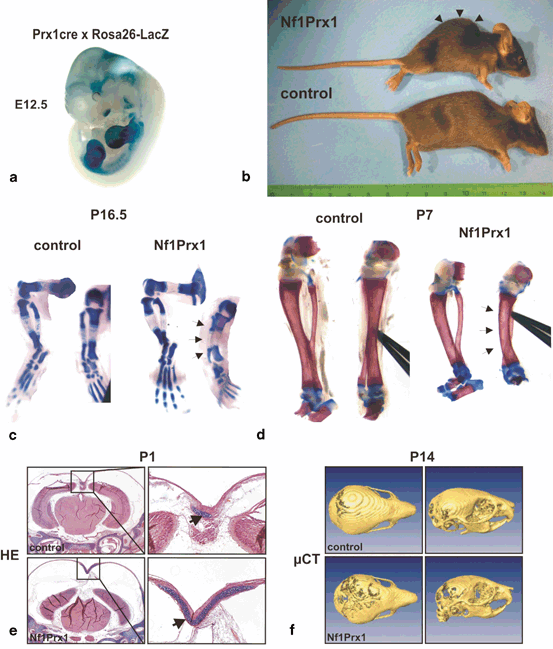
Fig. 2.3
Long bone dysplasia in neurofibromatosis type 1 (NF1). a Photograph of patient. b Radiograph of individual with NF1 with tibial bowing showing characteristic cortical thickening with medullary canal narrowing. c Radiograph of pseudarthrosis after long standing fracture of the tibia in an individual with NF1
Fig. 2.4
Gross morphology and skeleton in Nf1Prx1−/− mice. a Whole mount detection of the Cre recombinase activity in the cross of Rosa26-LacZ indicator mouse with Prx1-Cre mice. Note LacZ activity in the limb buds and in the cranial region. b A side view of the control and Nf1Prx1 −XE mutant mouse. Note that Nf1Prx1−mu mutant mice show a profound kyphosis (arrow heads), a phenotype often presented by mice featuring muscle dystrophy. c, d Tibia bowing in the prenatal and postnatal development in Nf1Prx1−Ti mice. Alcian blue and Alizarin red stained skeletal preparations of E16.5 and P7 mice in side and front view. Note a bowed tibial bone (arrows) visible in the front view of the tibia both at E16.7 and P7 stage indicating a prenatal origin of the phenotype. Note also shortening of the skeletal elements in the mutant mice. e, f Delayed mineralization and increased cartilage formation in the Nf1Prx1−De cranium. e Hematoxilin-Eosin/Alcian Blue stained transverse sections of the P1 wt and Nf1Prx1−Bl calvaria. Right panel shows magnification of the sagittal suture with enlarged cartilage area in the Nf1Prx1−al mouse calvaria (arrows). f Delayed mineralization of the calvarial bones in the Nf1Prx1 −XE mice visualized by µCT scan. The top view and the profile view of the scull, with poorly mineralized, transparent areas in the hind cranium of 14 days old Nf1Prx1 −XE mouse
Table 2.1
Callus cellular and structural characteristics in Nf1 mouse models. (Updated and adapted from Stevenson et al. [39])
Nf1 +/- Ref [49] | Nf1 ob-/- | Nf1 Prx-/- Ref [47] | Nf1 ob-/- Ref [25] | NF1Col2-/- Ref [11] | Nf1(flox/-) AdCre Ref [36] | |
|---|---|---|---|---|---|---|
Nf1 genetic setting | +/- (global) | -/- in mature osteoblasts only | -/- in limb mesenchymal osteoprogenitors and progeny (chondrocytes, osteoblasts, endothelial cells, adipocytes, muscles) | +/- (global) -/- in mature osteoblasts only | -/- in mesenchymal osteoprogenitors and progeny (chondrocytes, osteoblasts) | +/- (global) -/- in differentiating callus cells including chondrocytes and osteoblasts |
Osteoclast differentiation | Increased (following ovariectomy) | Increased (osteoblast-dependent) | Increased (mechanism ND) | Increased | Increased | Increased |
Osteoblast differentiation | Decreased | Normal | Decreased | Decreased | Decreased | Decreased |
Mineralization | ND | Delayed | Delayed | Delayed | Delayed | Delayed |
Structural properties | Increased callus volume | Cartilaginous remnants | Reduced BV/TV | ND | Reduced closed and open fracture union rates | |
Reduced BV/TV | Reduced callus size | Increased fibrotic or undifferentiated tissue at fracture callus | ||||
Increased cortical porosity | Altered BV/TV | |||||
Cartilaginous remnants | Reduced BMD | |||||
Callus mechanical properties | ND | Decreased stiffness and maximum force | Reduced torsional stiffness | ND | Reduced vertebral stiffness, yield | ND |
force, peak force | ||||||
Reduced ultimate torque at failure | Reduced femoral rigidity, and bending strength | |||||
Callus marrow | Fibrocartilaginous tissue | Normal | Fibrocartilaginous tissue | ND | ND | Fibrocartilaginous tissue |
Growth Plate Disturbances
Nf1 deficiency in cartilage causes decreased chondrocyte proliferation and premature growth plate chondrocyte hypertrophy [10]. This is caused by decreased Indian Hedgehog (Ihh) expression in the growth plate and concomitant increase of p21/Waf1 expression that leads to reduced cartilage proliferation and dwarfism of the appendicular skeleton. Reduced Ihh expression is a direct cause of premature hypertrophy and short growth [10].
Nf1 deficient cartilage strongly expresses the Sox9 transcription factor, which is localized to the nucleus and is responsible for retention of cartilaginous properties. This persistent nuclear localization of Sox9 is observed in the Nf1Prx1−/− mice in the cartilage anlagen, but also in areas where it should not be expressed (e.g. during hip joint cavitation in joint cavity). Due to persistence of nuclear Sox9 expression, the hip joint cavity retains a cartilaginous phenotype and undergoes fusion with the pelvis [10]. Similarly cartilage is ectopically formed in other regions e.g. in the hind cranium (Fig. 2.4e). It has been proposed that the hip joint fusion phenotype is likely at least partially due to reduced muscle force, which normally initiates joint cavitation. However we note that cases of hip joint fusion have not been observed in individuals with NF1.
Both Nf1Prx1 −/− and Nf1Col2−/− mice show increased osteopontin (SPP1) expression in Nf1 deficient growth plate chondrocytes and osteoblasts [26]. The SPP1 gene encodes matrix bone sialoprotein (osteopontin), which is known to attract and stimulate osteoclasts [31, 32]. Thus, not only RANKL expression but also osteopontin expression explains increased numbers of osteoclasts observed in the primary spongiosa in NF1 deficient mice.
Disturbances in Muscle Mass
Similar to human NF1 tibial dysplasia cases, Nf1Prx1−/− mice show tibial bowing even at prenatal stages, suggesting an early embryonic defect in bone and skeletal musculature development. In postnatal stages, Nf1Prx1−/− mice show decreased skeletal muscle mass in extremities and profound developmental muscle dysplasia, characterized by a decrease of myotube number, increased of connective tissue volume, and increased number of split muscle fibers. This is primarily caused by a defect of late myoblast differentiation during embryogenesis (Fig. 2.5). While muscle progenitor cell migration appears normal, myotube formation was inhibited by Nf1 ablation, which was associated with increased Ras-MEK1-Erk1/2 signaling and increased proliferation of connective tissue cells [33]. Shortening of the muscles may contribute to tibial bowing, as muscle mass in the lower extremities is unilaterally positioned and exerts force on the tibia which is similar to the force exerted by the chord on a bow. Disproportionate growth of skeletal elements together with shortening of the muscle mass helps explain the bowing phenotype.
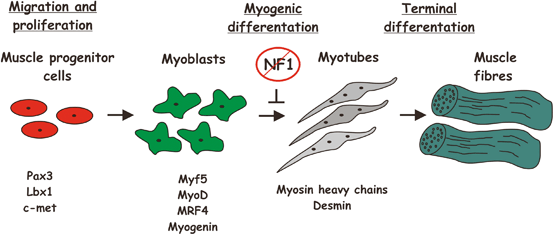
Fig. 2.5
Muscle development. (Adopted from Francis-West et al.; J. Anat. (2003)202, pp. 69–81). The regulation of primary myogenesis in the developing limb. The muscle precursors of the limb initially express the transcription factors Pax3 and Lbx1 together with the c-met tyrosine kinase receptor for scatter factor. Within the limb field, commitment to myogenic differentiation is marked by the expression of the myogenic regulatory genes (MRFs): Myf5 is detected first and is soon followed by MyoD. MRF4 is then transiently expressed and this is followed by myogenin expression. Subsequently, myoblasts terminally differentiate and express either slow or fast myosin heavy chain (MyHC) isoforms, which determine the muscle fibre type. The MyHC-expressing myoblasts fuse into multinucleate myotubes and assemble to form the muscle fibres. Loss of NF1 inhibits myoblast differentiation and myotue formation. (Reprinted from Journal of Anatomy, Francis-West PH, Antoni L, Anakwe K, Regulation of myogenic differentiation in the developing limb bud, p69–81, 2003, with permission from John Wiley and Sons)
Disturbances in Bone Structural and Material Properties
Bone structural properties are abnormal in NF1 . Bone homeostasis is controlled by an interplay of three main bone cell types: osteoblasts, osteocytes and osteoclasts. Osteocytes are the cells that arise at the end stage of osteoblast differentiation and are embedded deep in the mineralized bone matrix, communicating with one another through a network of canaliculi. In both the Nf1Prx1 −/− mouse and NF1 patient cortical bone samples, where functional neurofibromin is confirmed or presumed absent, osteocyte lacunae size is significantly increased (unpublished results—Dr. Jirko Kuehnich). Given that osteocytes constitute ≈90 % of living cells in the cortical bone, these enlarged osteocyte lacuna cause significant increase of bone microporosity that may contribute to structural weakening of the NF1 deficient bone. Also important is macroporosity due to bone demineralization in the proximity of blood vessels. Adult Nf1Prx1−/− mice have several-fold increased cortical bone macroporosity associated with the presence of large areas of demineralisation in the proximity of blood vessels penetrating the bone cortex. In mice, ectopic blood vessels were observed in the apex of the bone curve, which seem to predetermine the potential fracture sites. The bone surrounding the blood vessels is likely the primary site of the macroporotic bone structural destabilization in Nf1Prx1−/− mice (our unpublished observation).
Bone material properties are also abnormal upon loss of neurofibromin. Changes in bone material and bone structure likely play an important role in the etiology of NF1 long bone bowing. Nf1Prx1 −/− mice display softer bone as measured by nanoindentation experiments and contain less calcium than wild-type littermates [10]. In postnatal bones we detected lower bone mineral density and disorganized collagen fiber orientation; consistent with the idea that a significant problem in NF1 is due to defective extracellular matrix (ECM) maturation secondary to osteoblast dysfunction. Delayed osteoblast differentiation and the apparent inability to synthesize mature collagen, especially in the areas of high mechanical load (mid shaft) and in proximity of blood vessels cause focal bone lesions within cortical bone (unpublished results—Dr. Jirko Kuehnisch). A generalized problem is also observed in the form of lower bone mineral content in adult Nf1Prx1−/− mice. Based on currently available data, the defects of organic matrix maturation and mineralization are important in the etiology of long bone bowing. Thus, multiple converging defects of bone architecture as well as bone organic and mineral phase are responsible for the impairment of bone strength in NF1.
Disturbances in Blood Vessel Properties
Vascular anomalies have been reported in humans with NF1 (e.g. renal artery stenosis), and analysis of the blood from NF1 patients without vascular disease showed increased concentrations of inflammatory cells and cytokines linked to vascular inflammation [34]. Mouse models also support a role of neurofibromin in blood vessel properties as they are altered in Nf1 deficient mice. Nf1 +/− mice were shown to have increased neointima formation (i.e. thickened layer) after vascular injury [34]. This was associated with the vascular inflammation. It has also been shown that heterozygous inactivation of Nf1 in bone marrow derived cells, known to be involved in blood vessel homeostasis, is necessary and sufficient for neointima formation after vascular injury [34]. Thus, bone marrow cells appear to be involved in the inflammatory response in Nf1 deficient blood vessels, likely including vessels within cortical bone.
NF1 in Bone Regeneration
We now understand that neurofibromin is required for normal bone development as well as bone regeneration, but with an important distinction. During development and regeneration, loss of Nf1 has opposite effects in the early and late osteoblast differentiation. Inactivation in early osteo-progenitor cells inhibits osteoblast differentiation, triggers fibrous tissue formation (in context of fracture repair) and diminishes matrix mineralization. This is the case in Nf1Prx1 −/− mice where inactivation occurs in early limb bud mesenchyme and Nf1Col2−/− mice where inactivation takes place in early osteochondro-progenitor cells. In contrast, Nf1 inactivation at the osteoblast stage (e.g. in Nf1Col1−/− mice) leads to increased collagen synthesis and a net increase of bone formation. Thus Nf1Col1−/− mice exhibit both normal development and normal bone regeneration; closed fractures result in normal cartilaginous callus formation and formation of an enlarged, osteoid rich, bony callus [35]. However despite an enlarged callus, the bridging cortices are porous and overall fracture repair is delayed, indicating that late Nf1 inactivation interferes with normal healing processes. Moreover this model does not recapitulate features of tibal dysplasia-associated pseudarthrosis, and the fractures eventually heal.
Related posts:
 Insights into the Genetics of Clubfoot
The Genetic Architecture of Idiopathic Scoliosis
Molecular Genetics of Congenital Multiple Large Joint Dislocation
Overview of Next Generation, High-Throughput Molecular Genetic Methods
DMP-1 in Postnatal Bone Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column
Insights into the Genetics of Clubfoot
The Genetic Architecture of Idiopathic Scoliosis
Molecular Genetics of Congenital Multiple Large Joint Dislocation
Overview of Next Generation, High-Throughput Molecular Genetic Methods
DMP-1 in Postnatal Bone Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


