Fig. 5.1
Patient with idiopathic scoliosis. Curvature of the spine to the right with should imbalance is evident in the picture on the left. Standing postero-anterior spinal radiograph reveals severe thoracic curvature measuring > 70° by the Cobb angle method
Adolescent idiopathic scoliosis is coincident with the pre-pubertal growth spurt and affects ~ 2 % of the pediatric population, or about 29 million children worldwide (Fig. 5.2). Most AIS patients have non-progressive curves; that is to say, the deformity will not worsen appreciably. About 0.4 % of these children however will require active treatment to control progression, usually by bracing or surgery [3]. If left untreated, the natural history of progressive AIS is possible chest wall compromise with concomitant lung restriction, pain, deformity, and possible spinal osteoarthritis [1, 3]. Risk factors for progression in an affected child are well-documented. Female patients, and skeletally immature patients who present with large curves have a greater risk of progression, but additional predictive markers are highly sought [3]. Certain curve patterns, e.g. in the thoracic region, are also more likely to progress in severity and therefore must be monitored carefully. The potential for rapid, progressive deformity is significant enough that many professional health organizations recommend school screening programs, although their efficacy remains controversial [5]. The child who has AIS and is at risk of progression typically will be treated by bracing, with the goal of slowing or halting progression of the deformity. Although there is some evidence that bracing can be effective [3, 6], some deformities will continue to worsen, warranting surgery. Today surgery typically involves fixation with instrumentation and spinal fusion [2]. Although outcomes and safety in AIS treatment have benefited from recent advances in surgical and bracing techniques, the methods conceptually have remained the same for decades [3, 6].
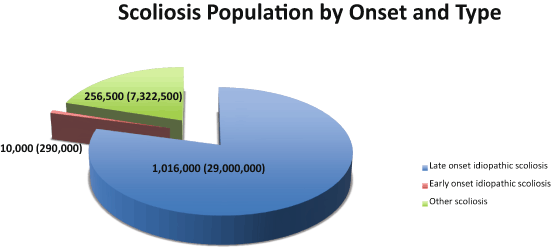
Fig. 5.2
Scoliosis populations. Pie chart depicts type of scoliosis (idiopathic shown in blue and brown, congenital shown in green) and proportion of each. The estimated numbers of scoliosis patients are given for the United States, with worldwide estimates in parentheses
As described in this chapter, although AIS is genetically heterogeneous, it bears several distinct features. For one, the location of spinal deformity in AIS patients (unlike infantile IS or syndromic scoliosis) usually involves vertebrae in the thoracic region [7]. In fact curve patterns in AIS appear to mirror spinal growth at this particular developmental stage in humans, which is known to be at its most rapid, particularly in the thoracic region [8]. Remarkably, greater than 90 % of thoracic curves are right-sided [2]. Girls are much more likely to have progressive deformity, leading some to the notion that AIS is a “female disease”. It is important to emphasize, however, that males comprise ~ 12–17 % of progressive cases [9]. Perhaps the most notorious of these was King Richard III of England, described by his acquaintance Thomas More as having “….croke backed, his left shoulder much higher than his right….”, a description borne out as scoliosis by his recently unearthed skeleton [10].
Less invasive interventions to halt or prevent scoliotic deformity altogether are clearly desirable. However until recently, etiologic understanding of the disease has lagged despite decades of clinical research. The reasons for this center on the complexity of the problem: the architecture of the spine itself (is AIS a problem of muscle, nerve, bone, or connective tissues?), the complex genetic underpinnings of AIS as described below, and lack of genetically-defined animal models that faithfully recapitulate the AIS phenotype. Recent advances in genomic technologies are fortunately proving to be powerful tools for deconstructing the causes of AIS in human patients. As outlined in this chapter, recent genetic discoveries have yielded new insights into AIS disease mechanisms. Here we describe the genetic underpinnings of AIS, and the progress in understanding AIS pathogenesis derived from gene discovery research. We also discuss emerging genetic investigations of AIS in humans and animal models, and the prospects for future molecular interventions.
Epidemiology and Inheritance—How Common and “Genetic” is AIS?
The prevalence of AIS is similar across all the major ancestral groups (summarized in Table 5.1), although some early studies found otherwise, likely reflecting differences in disease definition or diagnostic screening methods (e.g. physical examination versus radiography) [11–17]. Intra- and inter-observer error of 5° is generally accepted for measuring the Cobb angle, hence small curves are more likely to be false positives. If the standard scoliosis definition (> 10 degree Cobb angle method from standing lateral radiographs) is applied, most studies report prevalence of AIS in the range of 2–3 % of school age children. Consequently AIS does not generally cluster within any particular geographic region, nor is it evident that any population or ethnic group has been spared from AIS. Indeed, there is increasing evidence for genetic risk factors shared between ancestral groups as described later in this chapter.
Table 5.1
Published prevalence and heritability estimates for idiopathic scoliosis
(1) Country | Populations | Age group (years) | Minimum Cobb angle (degrees) | Prevalence | Inheritance (heritability) | Reference |
|---|---|---|---|---|---|---|
USA | Black, white | > 14 | > 10a | Black: 2.1 % White: 1.9 % | [11] | |
Canada | 12–14 | 5 10 | 4.5 % 2.0 % | [12] | ||
Sweden | ≥ 10 | Girls: 3.2 % Boys: 0.5 % | [13] | |||
England | 10–14 | ≥ 10 | 2.8 % | [16] | ||
Japan | Junior high school | ≥ 15 | Girls: 1.77 % Boys: 0.25 % | [14] | ||
Greece | 9–14 | ≥ 10 | 1.7 % | [15] | ||
USA | Boston (various ethnicities) | Families | Multifactorial | [22] | ||
USA | Utah | Families | > 10 | Mixed polygenic/recessive (96 %) | [23] | |
China | Chinese females | Families | ≥ 20 | Mixed polygenic/recessive (87.5 +/- 11.1 %) | [21] |
Genetic influences in AIS have been postulated for almost a century (reviewed in 7) [18]. Twin studies have consistently shown higher concordance in monozygotes compared to dizygotes, pointing to heritable factors [19, 20]. (Lack of full concordance in identical (monozygotic) twins reminds us of complex issues that are still poorly understood for genetic disorders, including reduced penetrance and possible environmental effects.) AIS sibling risk and heritability estimates also support significant genetic contributions. One study of AIS compared 415 Chinese index patients and found a sibling recurrence risk of 17.7 % and estimated a heritability of 87.5 % [21]. It is interesting to note that these results closely mirror an original recurrence risk estimate from a study of Caucasian cases 50 years prior [22]. A separate study of 100 U.S. Intermountain West probands with severe scoliosis (defined as requiring surgical correction) found an even higher recurrence risk of 33 % in first-degree female relatives. The latter study also suggested that the location of the scoliotic deformity on the spine may be more heritable than its pattern or severity [23]. At the population level, AIS inheritance is consistent with several to many mutations contributing greater than 80 % of disease risk [21–23]. However Mendelian (single gene) inheritance patterns including autosomal dominant, recessive, and X-linked dominant also have been described in AIS families [18, 24]. Accordingly both family- and population-based methods have been used in AIS gene discovery as described later in this chapter. Considering a multi- or poly-genic inheritance model, the preponderance of progressive AIS in females suggests that genetic loading may be greatest in affected males. In other words, males may require more risk mutations, or mutations with greater effect sizes, before they manifest disease. This phenomenon is known as the Carter effect [25], and recent data support this model in AIS inheritance [26]. It is also interesting to speculate that sexual dimorphism in AIS may be a function of gender-specific gene regulation, as has been described for other diseases such as liver cancer [27].
As with other complex disorders, there is considerable uncertainty in predicting individual genetic risk of developing AIS. For families with history of presumed AIS, care must be given to ensure that there is no underlying syndrome or known Mendelian disorder. Otherwise it is appropriate to advise patients with familial AIS that their recurrence risk is increased, but that quantifying the absolute risk is difficult [28].
AIS: A Disease of Bone, Muscle, or Nerve?
Neuropathologic mechanisms have long been proposed for AIS due to the association of scoliosis with neurologic/neuromuscular diseases. In laboratory animals, experimental models of scoliosis have been produced by surgically-created small brain lesions. One of the better-described methods involves removal of the pineal gland from chickens, fish, or bipedal rats, reportedly yielding scoliosis that resembles AIS [29–31]. Several investigations of AIS patients have suggested co-existing deficits in oculo-vestibular (visual/hearing) and proprioceptive function [32–36]. How primary alterations in the nervous system can effect spinal curvature is not altogether obvious, but presumably involves crosstalk with muscle/bone/connective tissue. Indeed we now understand that sensory innervation may affect bone homeostasis; for example, nerve-specific knockout of the axon guidance gene Sema3A produces low bone mass phenotypes in animal models [37]. It is interesting that several studies have found decreased bone mass in girls with AIS [38, 39]. One study found that osteopenia of the femoral neck was a prognostic indicator of curve progression, with an odds ratio of 2.3 [40]. Whether such a mechanism of nerve/bone crosstalk functions in skeletal growth and AIS will be important to consider in future hypothesis-driven studies. Other morphologic studies of AIS spinal structures have revealed some disarrangement of fibers of the ligamentum flavum in AIS patients compared to controls [41]; a separate study found decreased glycosaminoglycan content in the intervertebral discs of AIS patients [42]. Histochemical analyses of paraspinous muscles surrounding the scoliotic curve have shown relative hypertrophy and increased electromyographic signaling of type I fibers on the convexity of the curve in AIS patients. This was explained as most likely a compensatory response to curve progression [42, 43].
Identifying AIS Genes in Humans—Mapping Susceptibility Loci
AIS research in humans, consistent with most disease gene discovery efforts, has benefited from hypothesis-free, genome-wide methods that map the location of genetic risk factors in the genome relative to fixed markers [44]. One such method, linkage mapping, relies on identifying chromosomal loci that co-segregate with disease in families. Another method, association mapping, identifies polymorphic loci shared or transmitted more frequently to AIS cases (i.e. populations) compared to controls. Here we describe the results of both family-and population-based studies of AIS.
Family-Based Studies
As noted, both family- and population-based methods have identified genomic regions harboring putative AIS disease mutations. Table 5.2 lists five familial risk loci as given in the Online Mendelian Inheritance in Man (OMIM). Three loci, OS1, OS2, and OS5 (OMIM numbers 181800, 607354, and 612239) were mapped to chromosomes 19p13.3, 17p11, and 17q25-qter by linkage mapping in single East Asian (Han Chinese), European (Italian), and European (British) families [45–47]. Two additional loci IS3 (OMIM 608765), and IS4 (OMIM 612238) were each identified in family-based studies using independent cohorts and methods. IS3 was originally mapped to chromosome 8q12 by linkage mapping in a cohort of 52 families analyzed by model-free methods [48]. In similar fashion, IS4 was mapped to 9q31-q34 in a family with dominantly inherited AIS; moreover a suggestive linkage to this region was previously reported in a study of 202 affected sibling pair families [47, 49]. Consequently IS4 locus is potentially the first and only independently replicated AIS linkage. These studies underscore that AIS is genetically heterogeneous even between families. Indeed, one study suggested genetic heterogeneity even within a single family [50] (Table 5.2). One candidate gene, CHD7 encoding the chromodomain helicase DNA binding protein 7 transcription factor, has been proposed from linkage studies. Loss-of-function CHD7 mutations are well described in the CHARGE syndrome of multiple developmental anomalies that can include scoliosis [51, 52]. Tests of association produced significant results for common single nucleotide polymorphisms (SNPs) within a CHD7 intron, but how this gene region might be causal is unclear [48].
Table 5.2
Susceptibility loci in idiopathic scoliosis. Regions in the human genome providing statistically significant evidence of harboring an AIS risk factor are denoted by their OMIM locus number (column one) where applicable. The markers shown are those that provided most significant evidence in the region
Locus | Region | Marker | Mapping method | LOD score | P value (odds ratio) | Candidate gene | Reference |
|---|---|---|---|---|---|---|---|
IS1 | 19p13.3 | D19S216 | Genome-wide linkage in seven families | 3.63 | – | – | [45] |
IS2 | 17p11.2 | D17S799 | Genome-wide linkage in extended family | 3.20 | – | – | [46] |
IS3 | 8q12.1 | D8S1136 rs1038351 | Targeted linkage association across CHD7 | 2.77 | 0.0002 (GRR=3.1) | CHD7 | [48] |
IS4 | 9q31.2-34.2 | D9S2157 D9S915 | Genome-wide linkage in extended family genome-wide linkage in affected sibling pair familiesa | 3.64 – | 0.0005 | – – | |
IS5 | 17q25.3 | AAT095 | Genome-wide linkage in two families | 4.08 | – | – | [47] |
– | 5q13.3 3q12.1 | D5S2003 D3S2462 | Genome-wide linkage | 3.01 | – | – | |
– | 10q24.31 | rs11190870 | GWAS | – | 1.24×10−19 (1.56) | LBX1 | |
– | 6q24.1 | rs6570507 | GWAS | – | 1.27×10−14(1.27) | GPR126 |
Population-Based Studies
Genome-wide association studies (GWAS) typically involve genotyping a high density of common SNPs (≥ 350,000) that span the genome. Subsequent statistical comparisons of genotyped SNP allele frequencies between cases and controls may yield associated haplotypes that tag common disease loci. GWAS are proving effective for mapping common susceptibility loci in large cohorts of sporadic AIS. The Catalog of Published Genome-Wide Association Studies (http://www.genome.gov/gwastudies/) currently lists two studies (Table 5.2). The first GWAS of AIS utilized 419 families of various ethnicities, in a trio design that is robust to the effects of population substructure. This mostly non-Hispanic white cohort yielded strongest signals near the CHL1 gene encoding close homolog of L1, a cell adhesion protein involved in axon guidance [53]. The second study of 1033 East Asian (Japanese) cases and 1473 matched controls yielded strongest signals on chromosome 10q24.1 near the LBX1 gene. LBX1 encodes the Ladybird homeobox 1 protein involved in muscle and nerve specification [54]. This association was subsequently replicated in independent East Asian cohorts, and a larger combined analysis from multiple ethnic groups (i.e. mostly East Asian and non-Hispanic white) has provided further evidence for this susceptibility locus (Table 5.2). Thus, LBX1 is the first identified major AIS susceptibility locus. More recently, an expanded East Asian study yielded significant association with SNPs in the GPR126 gene encoding G-protein coupled receptor 126. This association was replicated in both East Asian and non-Hispanic white cohorts but is still untested in other ethnic groups. It is interesting that SNPs in GPR126 also have been associated with sitting height in humans [55]. Thus population studies have implicated several genes in multiple ethnic groups, although further study is clearly needed in under-represented minorities.
Chromosomal Breakpoint Mapping
Many chromosomal alterations with phenotypes that include scoliosis have been reported, although most do not appear to re-capitulate idiopathic forms, i.e., without obvious vertebral anomalies or co-existing diagnoses. One family segregating a pericentric inversion of chromosome 8 with idiopathic scoliosis has been reported [56]. Using methods of chromosomal breakpoint mapping, Bashiardes et al. found that one end of this inversion disrupted the 8q11.2 gene encoding gamma-1-syntrophin (SNTG1), while the other end of the inversion occurred in a gene-free region of 8p23. Subsequent analysis of the SNTG1 gene in 152 additional AIS patients revealed an apparent mutation in DNA samples from three unrelated patients. These changes were not detected in screens of 480 healthy control individuals. These results suggested that rare mutations in SNTG1 could occur in a small percentage of idiopathic scoliosis patients and left open the possibility that other nearby genes could be important in AIS. While chromosomal breakpoint mapping is still a powerful method for pinpointing disease genes, the majority of AIS cases are cytogenetically normal and not amenable to study by this method.
Candidate Genes and AIS Etiology
What have genetic mapping and association studies revealed about AIS biology? It is important to emphasize that, at this stage, candidate genes have been implicated because of their proximity to associated SNPs, rather than by any direct demonstrations of causality. Nevertheless, AIS candidate genes (Table 5.2) underscore potentially important disease pathways as illustrated in Fig. 5.3
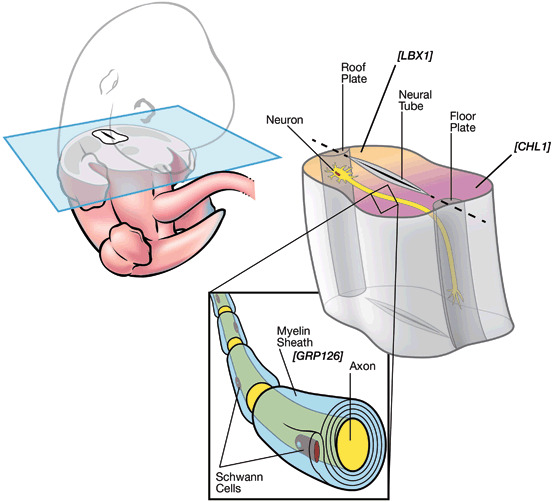
Fig. 5.3
AIS candidate genes and functions. A transverse section of developing spinal cord in mouse E12.5 is depicted. LBX1 and CHL1 are expressed in roof plate (shaded yellow) and floor plate (shaded magenta) of developing spinal cord, respectively. GPR126 is expressed in the myelin sheath surrounding axons
LBX1
LBX1 is the vertebrate ortholog of Ladybird Late (lbl) homeobox gene originally discovered in the fruitfly Drosophila melanogaster [57, 58]. In the fly, lbl participates in segmentation and cell specification of heart and muscle precursors. Targeted inactivation in the mouse has shown that this gene is necessary for lateral migration of muscle precursors into the limbs [59]. Further, loss of LBX1 causes loss of dorsal association neurons, cells of the dorsal spinal cord that relay somatosensory information, and disruption of dorsal horn innervation by nociceptive afferent neurons along with an increase in dorsal commissural neurons [60, 61].
Related posts:
 Insights into the Genetics of Clubfoot
Molecular Genetics of Congenital Multiple Large Joint Dislocation
Overview of Next Generation, High-Throughput Molecular Genetic Methods
Neurofibromin in Skeletal Development
DMP-1 in Postnatal Bone Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column
Insights into the Genetics of Clubfoot
Molecular Genetics of Congenital Multiple Large Joint Dislocation
Overview of Next Generation, High-Throughput Molecular Genetic Methods
Neurofibromin in Skeletal Development
DMP-1 in Postnatal Bone Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


