Fig. 4.1
Schematic structure of DMP1 protein, gene and ARHR mutrations. a Schematic structure of the human DMP1 gene and ARHR mutations. DMP1 is composed of six exons. Because of the recessive nature of this disease, there are only 7 variable mutations reported in the literature (see text for details). b Schematic structure of Full-length DMP1 protein:DMP1 signal peptide, MKTVILLVFLWGLSCAL, 37-kDa N-terminal fragment, a main cleavage site at peptide bond Ser196-Asp197, and the 57-kDa C-terminal fragment
Dmp1 Mutation/Deletion Leads to Autosomal Recessive Hypophosphatemia
DMP1 is mainly expressed in mineralized tissues and plays an important role during osteogenesis [13], although some in vivo animal studies showed that DMP1 may also have potential functions in non-mineralized tissues such as in brain [14], kidney [15] and salivary tissues [16]. For example, DMP1 is thought to actively promote and control the mineralization of collagen fibers and crystal growth within osteoids and predentin when these tissues are converted to mature bone and dentin [13]. In addition, DMP1 regulates and supports the maturation of osteoblasts to osteocytes and pre-odontoblasts to odontoblasts [17]. Importantly, DMP1 mutations in humans or deletion in mice lead to autosomal recessive hypophosphatemic rickets (ARHR)/osteomalacia [18]. It is noteworthy that, unlike other genetic disease cases in which human mutations are identified first and then animal studies such as gene knockouts are performed, it is the Dmp1 null mouse research results [19, 20] that triggered searches for DMP1 mutations in humans.
Autosomal Recessive Hypophosphatemia
Rickets is the softening of bones in children because of a deficiency or impaired metabolism of vitamin D, phosphorus, or calcium. This disease not only interrupts child development but also can potentially lead to fractures and disturbance of normal ossification. Rickets due to nutrition deficiency is more common in children in developing countries. The adult equivalent of rickets is known as “osteomalacia”, a disease characterized by inadequate or delayed mineralization of osteoids in mature cortical and spongy bones. Hypophosphatemic rickets/osteomalacia is a group of disorders characterized by hypophosphatemia, which mainly results from disorders of renal phosphate wasting (Unlike the situation with calcium, there is no rickets due to phosphorus deficiency, since one’s daily diet is rich in this element). There are several inherited gene mutations responsible for deficiencies in reabsorption in the renal tubule, which results in hypophosphatemia. For example, mutations in the PHEX gene (encoding phosphate regulating gene with homologies to endopeptidases on the X chromosome) lead to x-linked dominant hypophosphatemic (XLH) rickets [21], FGF23 (encoding fibroblast growth factor 23) mutations result in autosomal dominant hypophosphatemic rickets (ADHR) [22], and homozygous inactivating mutations in DMP1 cause autosomal recessive hypophosphatemic rickets (ARHR)/osteomalacia [18, 23 ,25]. Comparisons of patients having XLH, the most common hypophosphatemic rickets, there are fewer than ten cases of kindred ARHR have been identified worldwide due to the autosomal recessive nature of the DMP1 mutations [18, 23–26]. Molecular genetic analysis of these cases revealed the following changes: (1) the deletion of nucleotides 1484–1490 in exon 6 (1484–1490del) resulting in a frameshift that replaced the 18 conserved C-terminal residues with 33 unrelated residues, or another deletion in the same exon leading to a frameshift replacing 335 conserved amino acids with 53 unrelated ones; (2) a mutation in the start codon (1A→G) causing a methionine to valine change (M1V); (3) a homozygous 1-bp deletion in exon 6 (362delC) led to the placement of a premature stop codon after 120 unrelated amino acids; (4) a biallelic nucleotide substitution at nucleotide 98 in exon 3 (98G→A) introduced a premature termination codon (PTC) at codon 33, which replaced the wild type tryptophan residue (W33X); and (5) a mutation at the splice acceptor junction of exon 6 (IVS5 − 1G→A) or intron 2 (55 − 1G→G) is predicted to alter pre-mRNA splicing, which results in a shift in the open-reading frame if the final message is stable (Fig. 4.1b).
Considering the reported cases, the primary clinical symptoms of ARHR include: lower limb deformities (bowed legs or knock-knees), waddling gait, short stature or stunted growth, tooth abscesses or early loss of teeth, bone and muscle pain, biochemical abnormalities (hypophosphatemia with normal levels of serum calcium and parathyroid hormone). More severely afflicted patients may also suffer from nerve deafness, facial and dental abnormalities, learning disabilities, joint pain, and contracture and immobilization of the spine. Patients diagnosed with ARHR display symptoms in their early childhood that are likely to have a wide spectrum of severity, depending on the site and size of the mutations and the severity and chronicity of the associated phosphate depletion [27] (see Table 4.1).
Table 4.1
Characteristics of ARHP patients and mice models
Aspects | Characteristic features | |
|---|---|---|
Clinical | Figure | Short stature or growth retardation |
Posture | Waddling gait, immobilization of the spine, kyphosis | |
Craniofacial | Facial abnormalities, tooth abscesses or early loss | |
Limb | Genu varum or knock-knees | |
Symptom | Bone and muscle pain, joint pain, contracture, enthesopathy, nerve deafness, learning disabilities | |
Skeletal | Craniofacial | Enlarged dental pulp chambers and thin dentin, thick calvarium and skull base, skeletal malocclusion |
Spine | Disappearance of intervertebral disks and disk space, ossification of the longitudinal ligament | |
Joint | Loss of articular cartilage, narrowed joint space, osteophytes | |
Limb | Short, broad, bowing, ossification of tendon attachments, pathological bone fractures | |
Chest | Rachitic rosaries, narrow chest with wide clavicles | |
Biochemistries | Increase | FGF23, ALP |
Decrease | Pi, TmP/GFR | |
Normal | Ca | |
As with the human patients, Dmp1 null mice do not have apparent abnormalities during prenatal bone development, but severely impaired after birth presented as a chondrodysplasia-like phenotype that characterized by short and widened long bones with flared and irregular metaphyses, and malformed ossification centers with delayed development during postnatal growth [20]. In accordance with human patients, Dmp1 null animals display significantly lower serum phosphorus level than the wild type controls, which is due to the increased urinary phosphate excretion. In addition to human patients, Dmp1 null mice also significantly increase their serum PTH level [12, 17, 18]. Bone histological studies demonstrated that the Dmp1 null mice displayed severe osteomalacia, which show as a significant reduction in bone mineralization together with increased osteoid and more porous cortical bone, compared to wild type littermates. Under higher-magnification back-scattered scanning electron microscopic (SEM) images, the minerals, which are normally found to be evenly distributed around the osteocyte lacunae, were either absent or sparsely located in regions surrounding the Dmp1 null osteocytes. Scanning transmission electron microscopic images indicated a much lower content of mineral, calcium, and phosphorus in the Dmp1 null mineralized matrix (Fig. 4.2a, b, c) [12, 17, 18]. These observations suggested that genetic removal of Dmp1 (in mice) and loss-function of DMP1 mutation (in human ARHR kindreds) concurrently lead to independently altered skeletal mineralization and disturbed phosphate homeostasis .
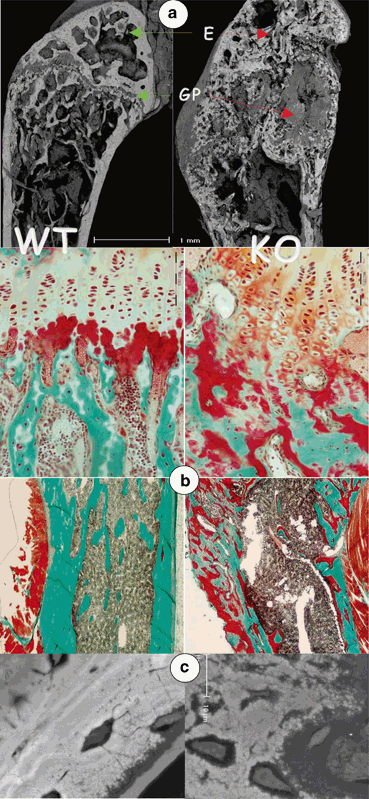
Fig. 4.2
Defective osteocytes are responsible for abnormal bone formation in the Dmp1 knockout (KO) mice (right panels). a Backscattered scanning electron microscopic (BSEM) images revealed the expanded and poorly organized growth plate (GP) in the Dmp1 KO femur, and severe defects in mineralization as reflected by discontinuous mineral content in the cortical bone region; b The Goldner stain showed sharp increases in osteoid areas (red) but greatly reduced mature bone (green) in the KO bone; and c BSEM images revealed that the mineral was evenly distributed surrounding the osteocyte lacunae in the control bone (left, white); however, the mineral content was either missing, or sparsely located in regions surrounding the Dmp1 KO osteocytes
Osteocyte Maturational Defects in Dmp1 Null Mice
An early transfection study of MC3T3-E1 cells that expressed excessive DMP1, displayed accelerated differentiation of osteoblasts and an earlier onset of mineralization, suggesting that DMP1 plays an important role during osteoblast differentiation, as well as osteocyte maturation and function [28]. It was later reported that patients with DMP1 mutations or mice with Dmp1 deletion did not display any gross abnormalities during the embryonic period or at birth [29]. However, they generally appeared to have the ARHP phenotype during their early postnatal development stage when DMP1 is highly produced by osteocytes and secreted into the mineral matrix. Further analysis indicated that Dmp1 deletion leads to defects primarily in osteocytes [12, 17, 18], cells that account for more than 95 % of bone cells.
The osteocytes reside in lacunae within the mineralized bone matrix and send their dendritic processes (ranging from 40 to 100 per cell) through tiny tunnels called “canaliculi” to form the osteocyte lacunocanalicular network [30]. As the osteocytes mature, numerous cellular projections form and elongate. Next, their cell volume and ultrastructure, such as the endoplasmic reticulum and the Golgi apparatus, are reduced, and a well-organized lacunocanalicular system is built up [30]. Compared to the well-established smooth inner wall of the lacunae and canaliculi, Dmp1 null osteocytes display bulky and coarse microstructural features, as well as abnormally enlarged and round-shaped osteocytes accompanied by a reduction in dendrite numbers [12, 17, 18]. A number of studies have consistently suggested the key pathological cause responsible for the ARHP phenotype in Dmp1 null mice is the maturational and functional defects of the osteocytes [12, 17, 18].
There is general agreement that dramatic decreases in protein expression and metabolic activity occur during the maturation of osteoblasts into osteocytes [31]. However, the analysis of Dmp1 null mice showed sharp increases of osteoblastic marker expression levels, such as runt-related transcription factor 2 (RunX2), osterix (OSX), Col I, alkaline phosphatase (ALP), bone sialoprotein (BSP), and osteocalcin (OCN). In addition, the early osteocyte marker E11/gp38 is widely and increasingly expressed throughout the whole cortical bone layer in the Dmp1 null mice. In contrast, sclerostin (SOST), mainly expressed in mature osteocytes, is greatly reduced. These studies indicated that osteocyte differentiation is likely regulated by DMP1 [12, 18, 19]. In addition, there is strong evidence showing that the osteocytes, the terminal differentiated cells, regain the ability to divide and proliferate in the Dmp1 mice [11, 12, 17, 18, 20] (see Fig. 4.3 for the working hypothesis).
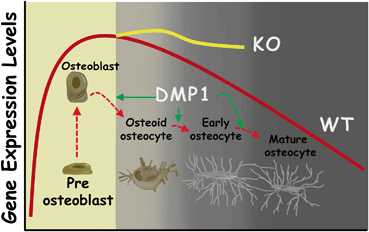
Fig. 4.3
Roles of DMP1 in control of osteocyte maturation. The gene expression levels are high during differentiation of osteoblasts from preosteoblasts, whereas the gene expression levels are gradually reduced during osteocyte maturation from osteoid osteocyte-early osteocyte and then mature osteocytes. DMP1 is one of key players during this process. The loss of Dmp1 in mice or DMP1 mutations in humans will disrupt normal osteoblast to osteocyte maturation. As a result, these immature osteoblast-like osteocytes express high levels of many osteoblast markers (RunX2, OSX, Col I, ALP, BSP, and OCN) and early osteocyte markers such as E11 in bone matrices
Dmp1 Regulates Osteocyte Biology and Bone Development
As previously mentioned, DMP1 is essential for osteogenesis by regulating the maturational process of osteoblasts to osteocytes and bone mineralization during postnatal development. As a key regulator, DMP1 performs several functions in osteocyte biology and bone development as described below.
DMP1 Directly Promotes Hydroxyapatite Formation In Vitro
The initial finding linking DMP1 with mineralization was based on the close association of the DMP1 expression and the in vitro “bone nodule” formation in primary rat calvarial cell cultures [4]. Soon after, He and colleagues reported that the specific acidic clusters in DMP1 molecules can provide the molecular design necessary for controlling the formation of oriented calcium phosphate crystals, and that the self-assembly of acidic clusters into a beta-sheet template of DMP1 is likely required for the role of DMP1 in biomineral induction [32]. Gajjeraman et al. also showed that both full-length recombinant DMP1 and native DMP1 C-terminal fragments isolated from rat bone accelerated the nucleation of hydroxyapatite in the presence of type I collagen, whereas the N-terminal domain of DMP1 (amino acid residues 1–334) inhibited hydroxyapatite nucleation [33]. Further analysis of these three DMP1 fragments within the mineralized tissues [N-terminal, C-terminal, and a chondroitin-sulfate-linked N-terminal fragment (DMP1-PG)] implies that both the C-terminal and N-terminal fragments are promoters of hydroxyapatite formation and growth, while DMP1-PG is an inhibitor [34]. These findings appear controversial although they indicate that distinct forms of DMP1 may work collectively to control the mineralization process in a different manner. However, the direct role of DMP1 in mineralization described above is mainly based on in vitro culture studies with little in vivo evidence.
DMP1 Signaling
DMP1 Signals via Cell Surface Integrin
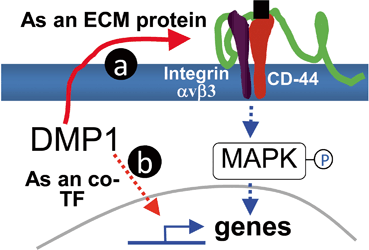
In vitro studies have shown that DMP1 promotes cell attachment through its RGD motif in a cell- and tissue-specific manner, suggesting a possible role for this protein in specific cell types to activate signaling pathways. This speculation is strengthened by the observation that exogenous DMP1 added to the exposed dental pulp may act as a morphogen trigger and/or promoter of the differentiation of undifferentiated ectomesenchymal cells in the pulp toward the odontoblast lineage. A further in vitro analysis demonstrated that either the full-length DMP1 or the 57-kDa fragment can activate phosphorylated extracellular signal-regulated kinase (p-ERK), with the effect of the 57-kDa fragment lasting longer than that of the full-length protein [35]. More recently, two separate studies confirmed that matrix DMP1 has the ability to activate the mitogen activated protein kinase (MAPK) pathways via interaction with cell surface ɑvβ3 integrin. This interaction stimulates the activation of its downstream effectors, namely as extracellular signal-related kinases (ERK1/2) and Jun N-terminal kinases (JNK1/2). These kinases later lead to the nuclear phosphorylation of c-Jun and ATF2, which shown as transcriptional factors that involved in regulating osteoblastic gene expression. [36, 37] (Fig. 4.4a) .
 Insights into the Genetics of Clubfoot
The Genetic Architecture of Idiopathic Scoliosis
Molecular Genetics of Congenital Multiple Large Joint Dislocation
Overview of Next Generation, High-Throughput Molecular Genetic Methods
Neurofibromin in Skeletal Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column
Insights into the Genetics of Clubfoot
The Genetic Architecture of Idiopathic Scoliosis
Molecular Genetics of Congenital Multiple Large Joint Dislocation
Overview of Next Generation, High-Throughput Molecular Genetic Methods
Neurofibromin in Skeletal Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column




Related posts:
Insights into the Genetics of Clubfoot
The Genetic Architecture of Idiopathic Scoliosis
Molecular Genetics of Congenital Multiple Large Joint Dislocation
Overview of Next Generation, High-Throughput Molecular Genetic Methods
Neurofibromin in Skeletal Development
Genetic and Environmental Interaction in Malformation of the Vertebral Column
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
