24 Prostaglandins, Leukotrienes, and Related Compounds
Eicosanoid biosynthesis is catalyzed by cyclooxygenases and lipoxygenases.
Eicosanoids and their receptors regulate inflammatory and immune responses.
Eicosanoid synthesis is modified by administration of precursor fatty acids.
The addition of oxygen to arachidonic acid and other polyunsaturated fatty acids not bound to membrane phospholipids in nearly all human cell types results in formation of several classes of bioactive products termed eicosanoids. These include prostaglandins (PGs), prostacyclin (PGI), thromboxanes (TXs), leukotrienes (LTs), and lipoxins (LXs). All of these compounds are crucial to the regulation of immunity and inflammation, among other physiologic and pathologic processes. Although eicosanoids are derived from C20 polyunsaturated fatty acids (eicosa = 20), only a small percentage of these polyenoic acids form the eicosanoids: dihomogamma linolenic acid (DGLA), which is 8,11,14-eicosatrienoic acid; arachidonic acid (AA), which is 5,8,11,14-eicosatetraenoic acid; and eicosapentaenoic acid (EPA), which is 5,8,11,14,17-EPA (Figure 24-1).
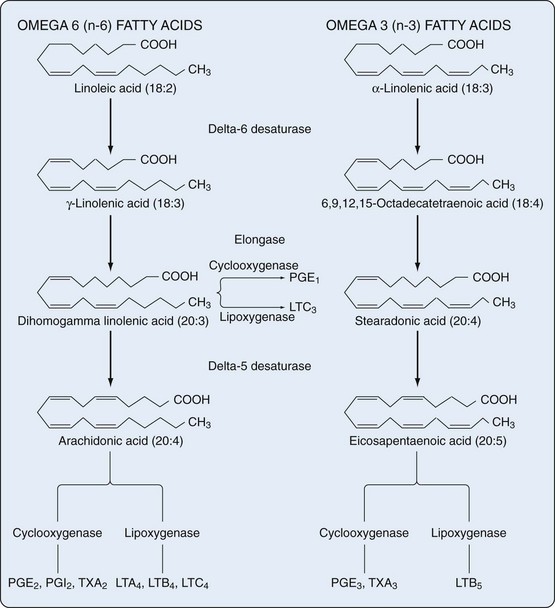
Two groups of fatty acids are essential to the body: the omega-6 series derived from linoleic acid (18:2 n-6) and the omega-3 series derived from α-linolenic acid (18:3 n-3). The n refers to the number of carbon atoms from the methyl (omega) end of the fatty acid chain to the first double bond (i.e., omega-3 and omega-6 designations). Using this notation, 18 refers to the number of carbon atoms in the fatty acid. The degree of unsaturation (the number of double carbon-carbon bonds) follows the number of carbon atoms. Fatty acids are metabolized by an alternating sequence of desaturation (i.e., removal of two hydrogens) and elongation (i.e., addition of two carbons). Membrane phospholipids are the main storage site for polyunsaturated fatty acids and are particularly rich in eicosanoid precursors, which are located at the sn-2 position (Figure 24-2). Because mammalian cells cannot interconvert n-3 and n-6 fatty acids, the composition of membrane phospholipids is determined by exogenous sources of fatty acids.
Biosynthesis of Eicosanoids
Phospholipases
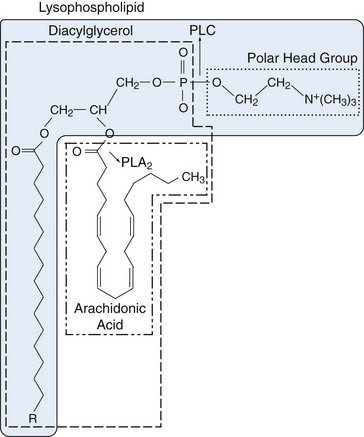
Phospholipase A2 (PLA2) in lysosomes or bound to cell membranes catalyzes the breaking of the sn-2 bond, facilitating release of AA or other polyunsaturated fatty acids (see Figure 24-2). The enzyme is crucial to regulation of eicosanoid synthesis because it is in the nonesterified state that the polyunsaturated precursors enter into the cascades, leading to eicosanoid formation. Only a scant amount of oxidation at carbon 15 of AA occurs catalytically when the fatty acid is still covalently bound as part of a phospholipid.1 More than 25 different PLA2 isoforms have been characterized and grouped based on primary structure, localization, and Ca2+ requirements.2 Cytosolic PLA2 group IV is the major catalyst of arachidonate release that leads to PG and LT production. The structure of cPLA2 reveals a monomeric cytosolic protein with a molecular size of 85 kD. Potent and selective inhibitors of cPLA2 have been developed.3 Lysophospholipids “left over” after the action of PLA2 are direct precursors of platelet-activating factor (PAF), a potent mediator of inflammation, which is generated by acylation (addition of fatty acid) in the open sn-2 position of the lysophospholipid. One member of the PLA2 superfamily, lipoprotein PLA2, may serve as a marker for cardiovascular risk.4
Four distinct types of PLA2 activities hydrolyze fatty acids esterified at the sn-2 position: Secretory PLA2 (sPLA2) has small disulfide cross-linked proteins that require Ca2+ in mM concentrations for optimal activity. Cytoplasmic PLA2 (cPLA2) has larger proteins requiring Ca2+ in µM concentrations, which are AA-selective and can deacylate diacylphospholipids completely, preventing accumulation of potentially toxic lysophospholipids. Ca2+-independent PLA2 (iPLA2) exhibits specificity for plasmalogen substrates. Finally, PAF acetylhydrolase (PAF-PLA2) has a series of isozymes specific for short chains.4
Under basal conditions, AA is liberated by iPLA2 and then reincorporated into cell membranes (reacylated). It is unavailable for appreciable eicosanoid biosynthesis. The acylase enzymes competitively inhibit the cyclooxygenase (COX) isoenzymes. After receptor activation and cell stimulation, intracellular Ca2+ levels increase and the Ca2+-dependent cPLA2 liberates arachidonate at a rate that exceeds the rate of reacylation, leading to arachidonate metabolism by COX isoenzymes and by lipoxygenases (LO). Initial formation of PGE2 seems to be due to preferential coupling among cPLA2, COX-1, and cytosolic PGH-PGE isomerase. More intense inflammation leads to participation of secreted sPLA2 and amplification of PGE2 biosynthesis by inducible COX-2. It is an oversimplification to regard availability of AA as the sole rate-limiting step in cellular eicosanoid biosynthesis. The coordinated action of phospholipases and the restricted expression and altered activation of COX isoenzymes are also important.5
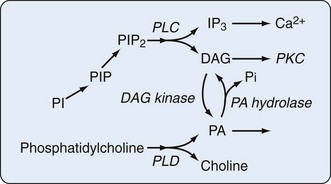
Phospholipase C (PLC) hydrolyzes the polar head group (e.g., inositol, choline) from phospholipids to yield diacylglycerol (DAG) and the polar head group. Direct protein isolation and molecular-cloning studies have revealed multiple PLC isozymes in mammalian tissues. Phosphatidylinositol-PLC occurs in cytosolic (cPLC) and secreted (sPLC) forms and can be divided into three major classes (PLC-β, PLC-γ, and PLC-δ) based on substrate specificity. PLC with specificity for phosphatidylinositol and phosphorylated phosphatidylinositol is a key component of phosphatidylinositol-mediated signaling pathways. DAG is an activator of protein kinase C (PKC), and rapid production of this lipid by phosphatidylinositol-PLC hydrolysis of the phosphorylated phosphatidylinositol pool is a primary step in signaling. Further AA is made available by the sequential actions of diglyceride lipase and monoglyceride lipase.6 PLC with activity on phosphatidylcholine has also been identified. Peripheral blood monocytes from patients with rheumatoid arthritis (RA) exhibit greater PLA2 and PLC activity than cells from healthy volunteers. PLA2 concentrations did not correlate with disease activity, but the greatest increases in PLC enzyme activity were seen in cells from patients with the most severe, persistent, proliferative disease, not in cells from patients with the most active disease at the time the cells were studied.7 In accord with the need for balance in regulation of the inflammatory response, sPLA2, numbering nine members in humans, has both inflammatory and anti-inflammatory actions. Thus, group V sPLA2 counters the inflammatory activity of group IIA sPLA2 by regulating cysteinyl leukotriene synthesis and promoting immune complex clearance in a murine model of autoimmune erosive inflammatory arthritis. It is of interest that concentrations of IIA sPLA2 are far greater than those of V sPLA2 in synovial fluid from patients with RA.8 In keeping with the fact that nothing in biology and the pathogenesis of disease is simple is the observation that treatment of RA patients with sPLA(2)-IIA inhibitors has only transient benefit and that sPLA(2)-IIA has activity (induction of COX-2) independent of its enzyme activity.9
Hydrolysis of phospholipids by phospholipase D (PLD) produces phosphatidic acid (PA) and the respective polar head groups. The capacity of cells to interconvert PA and DAG through the action of specific cellular phosphatases and kinases (Figure 24-3) suggests that AA release from DAG and a variety of intracellular signaling and protein-trafficking events may be regulated by PLD activity. The targets of phosphorylation within the PLD2 molecule that are key to its regulation have been mapped. PLD may be activated after or independent of PLC activation.10
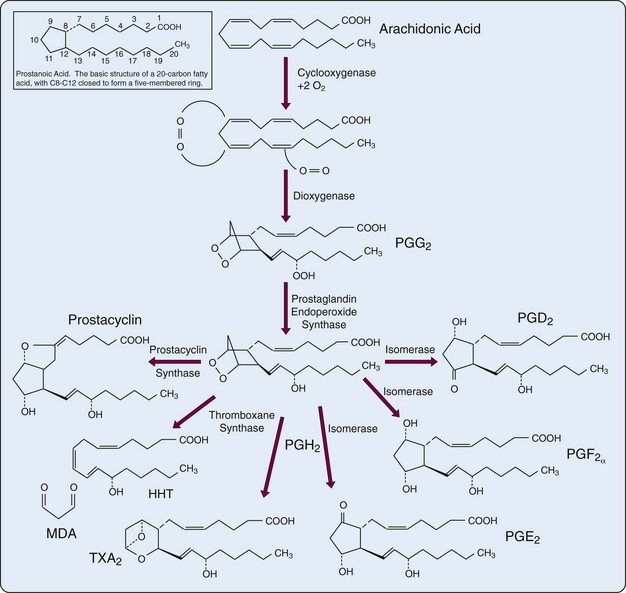
The tetraenoic precursor (AA) is the most abundant of the three precursor fatty acids in cells of individuals who eat usual Western-style diets. Metabolites of AA constitute the “2” series (dienoic) PGs (two double bonds in the molecule), and the metabolic pathway has acquired the familiar name “arachidonic acid AA cascade.” However, diets enriched in eicosapentaenoic or gamma-linolenic acid, the other eicosanoid precursors, lead to formation of different eicosanoids. Figure 24-4 illustrates the COX and 5-LO lipoxygenase pathways of the cascade.
Cyclooxygenase Pathway
The first step in the biosynthesis of the “prostanoids” (e.g., PGs, TXs, prostacyclin) is catalyzed by the bifunctional PG endoperoxide synthase isozyme (PGHS)-1 (COX-1) and PGHS-2 (COX-2). COX is a homodimeric enzyme that integrates into only a single leaflet of the lipid bilayer of the cell membrane. The COX active site is a long hydrophobic channel. Aspirin and most other nonsteroidal anti-inflammatory drugs (NSAIDs) exclude arachidonate from the upper portion of the channel.11 To form the characteristic five-carbon ring structure (TXs contain a six-member ring), the precursor fatty acids must have double bonds at carbons 8, 11, and 14 (numbering from the carboxyl group). When a molecule of oxygen is inserted across carbons 9 and 11, ring closure occurs enzymatically across C8 and C12, creating the unstable PG endoperoxide PGG. Subsequent peroxidation yields PGH with formation of the cyclopentane ring. PGH serves as the common precursor for PGs, prostacyclin, and TXs that are formed under the influence of terminal synthases (see Figure 24-4). In addition to the activity of phospholipases, regulation of PG synthesis also occurs at the level of PGHS gene expression. PGHS levels are increased by interleukin (IL)-1, platelet-derived growth factor, and epidermal growth factor, agents that increase PG synthesis.
Cell membranes constitute the source of substrate AA and the site of action of eicosanoid-forming enzymes. PG synthesis can also form at lipid bodies, which are non–membrane-bound, lipid-rich cytoplasmic inclusions that develop in cells associated with inflammation. Lipid bodies isolated from human monocytes express PGHS activity, are reservoirs of arachidonyl phospholipids, and can function as domains of PG synthesis during an inflammatory reaction.12
PGHS, the well-known target of NSAIDs, exists in two isoforms. These isoforms are similar in terms of amino acid identities (about 60%), catalytic properties, and substrate specificity, but they differ in their genomic regulation.13
Regulation of Cyclooxygenase-1 Expression
The gene for COX-1 (PTGS1) is preferentially expressed constitutively at high levels in selected cells including endothelium, monocytes, platelets, renal collecting tubules, and seminal vesicles. Because the expression level of the enzyme does not vary greatly, it has been difficult to study its transcriptional regulation. The gene has a TATA-less promoter that contains multiple start sites for transcription. It is also known that Sp1/Cis regulatory elements in the promoter bond the Sp1 transcription factor to induce COX-1 gene expression. In addition, COX-1 (COX-2) splice variants may function in tissue-specific normal and pathologic processes and may represent new targets for therapy.14 The localization of COX-1 in nearly all tissues under basal conditions suggests that its major function is to provide eicosanoids for physiologic regulation. This is seen clearly in platelets that do not have nuclei and cannot produce an inducible enzyme on activation. Rather, TXs are produced constitutively so that platelet aggregation can be completed.
Regulation of Cyclooxygenase-2 Expression
The regulated formation of eicosanoids implies that cells have an appreciable ability to amplify the rate and amount of eicosanoid synthesis. Several processes contribute to that regulation including silencing of sPLA2 expression by COX-2 and autoinactivation (“suicide inactivation”) of COX-2 and other oxygenases and synthases. In addition, the COX-2 transcript contains at least 12 copies of the AUUUA RNA motif, which makes it unstable and subject to rapid degradation. Factors that regulate COX-2 expression are specific for the physiologic processes involved. Expression of COX-2 in the macula densa in the kidney depends on luminal salt concentrations. Transcriptional activation of the COX-2 gene by mediators of inflammation such as IL-1β and tumor necrosis factor (TNF) is likely regulated by the transcription factors nuclear factor κB (NFκB) and C/EBP. Perhaps the most crucial of the several demonstrated regulatory sequences in the 5′ flanking regions of the COX-2 gene is the ATF/CRE, a site that is activated by the transcriptional activator protein-1 (AP-1) and the cyclic adenosine monophosphate (cAMP) regulatory binding protein. Now that quantitative polymerase chain reaction analysis is routinely used to examine gene expression, it is clear that COX-2 is constitutively expressed under basal conditions at levels too low to be detected by Northern blot analysis.15 Depending on cell and tissue specificity, several signaling pathways (kinases, Rho, cyclic guanosine monophosphate, Wnt) and transcription factors (NFκB, AP-1, nuclear factor of activated T cells [NFAT]) are involved in COX-2 expression.16
COX-1 and COX-2 effect a balance in several physiologic and pathologic situations. Of particular interest are their actions in kidney and stomach. During times of low blood volume, the kidney releases angiotensin and other factors to maintain blood pressure by systemic vasoconstriction. Angiotensin also provokes PG synthesis in the kidney. COX-1, expressed in vessels, glomeruli, and collecting ducts, produces vasodilating PGs, which maintain renal plasma flow and glomerular filtration during conditions of systemic vasoconstriction. In the antrum of the stomach, COX-1 leads to production of PGs, which increase gastric blood flow and mucus secretion. Inhibition of COX-1 by NSAIDs prevents these protective mechanisms and results in renal ischemia and damage and gastric ulcers (mainly antral) in susceptible individuals. These observations have led to development of NSAIDs that selectively inhibit COX-2 and spare COX-1. AA gains access to the active site of the COX via a hydrophobic channel, and access is blocked by insertion of an acetyl residue on Serine 530 in COX-1 and Serine 516 in COX-2. The irreversibility of the interaction and the unique expression of COX-1 in the anucleate platelet is the reason for the clinical efficacy of low-dose aspirin. Nonacetylated NSAIDs compete with arachidonate for the active site and can interfere with the sustained effects of aspirin. Although the structures of COX isozymes are similar, COX-2 is characterized by a side-pocket extension to the hydrophobic channel, which is where the selective COX-2 inhibitors localize.13
The major adverse effects of NSAIDs, gastroduodenal injury and impaired renal function, as well as the induction of myocardial infarction and stroke, are caused by inhibition of COX-1, whereas the analgesic and anti-inflammatory activities of NSAIDs rest in part on their ability to inhibit COX-2. COX-2 also seems to have a regulating role, however, in renal, brain, gastrointestinal, ovarian, and bone function. COX-2 is also expressed in endothelial cells, and its inhibition suppresses prostacyclin synthesis by endothelial cells.17 COX-2 acts in the initiation and the resolution of inflammation. Its expression increases transiently early in the course of carrageenan-induced pleurisy in rats. Later in the response, COX-2 is expressed at even higher levels, leading to synthesis of PGD2 and its dehydration product 15-deoxy-δ12,14-PGJ2 (15δPGJ2). Early expression of COX-2 is associated with production of inflammatory PGs, whereas the later peak results in production of PGs that suppress inflammation.18 That inflammation occurs in COX-2 knockout mice19 reminds us, as Lewis Thomas stated,20 “inflammation will take place at any cost.” Nonetheless, the search for more selective and localized COX-2 inhibition goes on. One strategy is achieved by using the mechanism of RNA interference (RNAi). Nonpathogenic Escherichia coli are engineered to invade tumor cells (and perhaps could be designed to invade synovial cells) and to generate anti-COX-2 siRNA molecules (siCOX-2), thereby silencing overexpressed COX-2. The involvement of miRNAs in COX-2 posttranscriptional regulation suggests a possible endogenous silencing mechanism to reduce COX-2 expression.21
Cyclooxygenase-3
Acetaminophen, similar to NSAIDs, suppresses pain and fever. It is not an anti-inflammatory agent, and its mechanism of action—despite its extensive use—has not been apparent. The finding that acetaminophen inhibits COX activity more in canine brain homogenates than in spleen homogenates gave rise to the concept that variants of the COX enzyme exist that are differentially sensitive to acetaminophen. COX-3 is a splice-variant of COX-1 that retains the intron-1 gene sequence at the mRNA level, which encodes a 30 amino acid sequence inserted into the N-terminal hydrophobic signal peptide of the enzyme protein. COX-3 protein and mRNA transcripts have been identified in human tissues as well as rats, most abundantly in cerebral cortex and heart, although the mechanism for conversion of COX-3 mRNA to active enzyme is not clear.11,22 These findings led to a search for other COX variants. A COX-2 variant induced by 0.5 mM diclofenac via stimulation of the nuclear receptor peroxisome proliferator-activated receptor (PPARγ) (the receptor for the pro-resolving 15-deoxy-δ12, 14 PGJ2) has been discovered. Whereas induction of COX-2 by LPS results in release of the inflammatory cytokines IL-6 and TNF, diclofenac-induced COX 2 leads to release of the anti-inflammatory cytokines TGF beta and IL-10.
Prostaglandin Synthases
Conversion of the endoperoxide intermediate PGH2 to PGs requires the activity of specific terminal synthases: Hematopoietic PGD synthase (H-PGDS) catalyzes the isomerization of PGH2 to PGD2 in immune and inflammatory cells. Cytosolic PGE synthase (cPGES) is responsible for constitutive expression of PGE2, and microsomal PGE synthase (mPGES-1) induces PGE in response to inflammatory stimuli. At least 10 enzymes convert PG precursors into biologically active PGs.23 Suppression of PG synthase activity might be considered as an alternative strategy that would fall between global blockade by inhibition of COX and blockade of a single eicosanoid receptor.24 Increased fibrosis may now be added to the well-known adverse effects of NSAIDs including COX-2 selective agents.25 Thus efforts have been directed at development of drugs that inhibit mPGES-1, rather than COX-2, thereby suppressing PGE2 production while sparing production of prostacyclin.3 Microsomal PGES-1 is upregulated throughout development of the rat adjuvant-induced arthritis model.26 Because PGE2 is also important to induction of matrix metalloproteinases MMP-3 and MMP-13, suppression of mPGES-1 might counteract articular cartilage degradation in patients with inflammatory arthritis.27
Products of the Cyclooxygenase Pathway
Prostaglandins
The basic structure of all PGs is a “prostanoic acid” skeleton, a 20-carbon fatty acid with a five-membered ring at C8 through C12 (see Figure 24-4, inset). The term prostaglandins is employed widely but should be used only to describe the oxygenation products that contain the five-membered carbon ring. A family of acidic lipids found first in human seminal fluid, PGs were misnamed because it was thought they were produced in the prostate gland rather than in the seminal vesicles.28–30 The alphabetic PG nomenclature (e.g., PGE, PGF, PGD) is related to the chemical architecture of the cyclopentane ring. PGE and PGF differ only in the presence of a ketone or hydroxyl function at C9 (see Figure 24-4). These compounds are made by a variety of cells (e.g., PGE2 and PGD2 by isomerases, PGF2α by a reductase). In the nomenclature a subscript numeral after the letters indicates the degree of unsaturation in the alkyl and carboxylic acid side chains. The numeral 1 indicates the presence of a double bond at C13-C14 (PGE1), 2 marks the presence of an additional double bond at C5-C6 (PGE2), and 3 denotes a third double bond at C17-C18 (PGE3).
PGs are produced on demand and seem to exert their effects on the cell of origin or nearby structures. They are not stored in cells and are degraded rapidly in vivo by 15-hydroxyprostaglandin dehydrogenase (PGDH) during one passage through the lungs. Abundant experimental evidence supports the view that PGs participate in development of the inflammatory response. Administration to rats of a monoclonal antibody to PGE2 prevents carrageenan-induced pain and inflammation.31 PGs are probably better at potentiating the effects of other mediators of inflammation than they are at inducing inflammation directly. PGE compounds and intermediate hydroperoxides of AA increase pain sensitivity to bradykinin and histamine. The effects of PGE are cumulative, depending on concentration and time. Even small amounts of PGs, if allowed to persist at the site of injury, may in time cause pain.
PGE2 stimulates bone resorption,32 and its 13,14-dihydro derivative is nearly as potent, which is of interest because derivatives of the biologically active PGs are usually assumed to be of no functional significance. Addition of serum to the culture medium stimulates bone resorption, a process that is complement dependent and PG mediated. The mechanism may help explain bone erosion in joints of patients with RA, in which complement is activated, and PGE2 concentrations are high. The observation that PGE1 can stimulate bone formation33 suggests that PGs physiologically participate in coordination of bone formation and resorption. For example, primary idiopathic hypertrophic osteoarthropathy, a familial disorder, is associated with mutations in PDGH and subsequent impairment of PG degradation. These patients have chronically elevated levels of PGE and exhibit digital clubbing and evidence of both increased bone formation and resorption in their phalanges.34 Many effects of IL-1 and TNF on cells are associated with stimulation of PG production and inflammation. Cartilage explants from osteoarthritis patients express COX-2 (but not COX-1) and release 50 times more PGE2 in culture than cartilage from healthy subjects and 18 times more PGE2 than normal cartilage stimulated with cytokines plus lipopolysaccharide. Explants from osteoarthritis patients release IL-1β, whereas normal cartilage does not express mRNA for pro-IL-1β or release IL-1β spontaneously. It seems that in osteoarthritis—and probably in RA—upregulation of cartilage IL-1β and subsequent production of PGE2 leads to cartilage degradation.35,36
Mast cells, often overlooked as important in inflammatory responses, are seen in large numbers in synovium from patients early in the course of RA.37 PGD2, the major PG formed by mast cells, also can mediate histamine release from mast cells exposed to anti-IgE antibody. PGJ2, formed from the dehydration of PGD2, seems to function as a brake on the inflammatory response. It reduces macrophage activation, reduces nitric oxide production from stimulated cells, and induces apoptosis in tumor cell lines. PGJ2 is metabolized to 15-deoxy-δ12,14 PGJ2 and δ12 PGJ2, which are also biologically active.38 It is not clear whether PGA, formed from loss of water from the cyclopentane ring, has biologic activity. However, in vitro, PGA2 induces apoptosis in HL-60 cells.39
Prostacyclin
Prostacyclin, discovered in 1976,40 has been purified, and the cDNA for prostacyclin synthase has been cloned. In addition to a cyclopentane ring, a second ring is formed by an oxygen bridge between carbons 6 and 9. It is generated from PGH2 by a distinct prostacyclin synthase, a 56-kD member of the cytochrome P-450 superfamily of enzymes found predominantly in endothelial and vascular smooth muscle cells.15 Production of prostacyclin can be stimulated by thrombin or generated by transfer of PGH2 from platelets (the endoperoxide steal), contact with activated leukocytes, or stretching of the arterial wall. It is a powerful vasodilator and inhibits platelet aggregation through activation of adenylate cyclase, which leads to an increase in intracellular cAMP. It is metabolized rapidly (half-life in plasma is less than one circulation time) to the more stable, less biologically active 6-keto-PGF1α. The enzymatic products of its conversion—2,3-dinor-6 keto-PGF1α and 6,15-di-keto-2,3-dinor PGF1α—are also chemically stable and have little biologic activity. They are the major metabolites of prostacyclin excreted in urine, in which they can be assayed as indicators of prostacyclin generation.
Prostacyclin generated in the vessel wall has antiplatelet and vasodilator actions, whereas TXA2 generated by platelets from the same precursors induces platelet aggregation and vasoconstriction. These two eicosanoids represent biologically opposite poles of a mechanism for regulating the interaction between platelets and the vessel wall and of formation of hemostatic plugs and intra-arterial thrombi. Given the central role of platelets in inflammatory reactions, an appropriate prostacyclin-TX balance is important to regulation of inflammation. The balance may be altered in patients with antiphospholipid antibody syndrome; in patients treated with cyclosporine; and in patients treated with NSAIDs, especially patients treated with selective COX-2 inhibitors. Although COX-2 inhibitors reduce recurrence of colorectal adenomas, they increase the risk for cardiovascular events such as myocardial infarction and stroke.41
Intravascular infusion of prostacyclin also reduces some of the clinical changes associated with pulmonary embolism. The instability of prostacyclin makes it cumbersome to administer therapeutically. Nonetheless, it has been used with limited success to treat peripheral vascular disease including Raynaud’s phenomenon. New therapeutic approaches for treatment of pulmonary hypertension including patients with sarcoidosis and systemic sclerosis are prostacyclin analogues such as epoprostenol, beraprost, and iloprost, but these are not free of adverse events due to intravenous administration.42,43 Several drugs including angiotensin-converting enzyme inhibitors and statins release endogenous prostacyclin from vascular endothelium in vivo, observations that pave the way for development of new anti-inflammatory and cardioprotective agents.44 In addition to its vasodilator effects, prostacyclin suppresses endothelial cell proliferation. Prostacyclin analogues might prove useful as adjunct treatment for some cancers.
Thromboxanes
The extraordinary rapidity with which platelets adhere to damaged tissue, aggregate, and release potent biologically active materials suggests that the platelet is well suited to be a cellular trigger for the inflammatory process. Efforts directed at suppression of TX synthesis and platelet aggregation may result in limitation of inflammatory responses, especially in coronary arteries. Inhibition of platelet aggregation may be important to the anti-inflammatory effects of aspirin and other NSAIDs. Long-term administration of low doses of aspirin (40 mg/day—the lowest dose predicted to cause total inhibition of TX formation in serum, according to mathematic modeling)—has inhibitory effects on platelet function ex vivo that are indistinguishable from the effects caused by giving 325 mg/day of aspirin.45 Aspirin acetylation of COX in platelets occurs in the portal vein where the aspirin concentration is high before it is metabolized in the liver, which explains why the 81-mg dose of aspirin is so effective in prevention of heart attacks and strokes. Platelets lose their ability to aggregate until new platelets are formed in about a week.11 Nonetheless, the response to aspirin can vary among individuals. It has been suggested46 that serum TXB2 levels be monitored to ensure the efficacy of aspirin therapy. Even low-dose aspirin has some depressive effect on prostacyclin synthesis, however. This means recovery of vascular prostacyclin production may be more difficult to attain in elderly patients.47
Selective inhibition of TX synthase represents an approach that may be used to suppress TXA2 synthesis without depressing prostacyclin formation. The endoperoxide steal seems to function in vivo after administration of a TX synthase inhibitor. Antagonists of the receptors shared by endoperoxide and TXA2 have been developed, and these agents inhibit platelet aggregation in patients who are recalcitrant to TX synthase inhibition. Novel agents targeting TX receptors and TX synthase have improved treatment of vasculitis and cardiovascular and renal diseases.48 More specific inhibition of TX action may become possible now that TX receptors have been cloned and characterized.49
Lipoxygenase Pathways
The human 5-lipoxygenase (5-LO) gene has been isolated and characterized50,51 and produces a 78-kD enzyme. In myeloid cells the 5-lipoxygenase pathway leads to formation of the biologically active LTs (Figure 24-5) that were originally found in leukocytes and that contain three conjugated double bonds (trienes). Cell activation leads to translocation of 5-lipoxygenase from cytosol to the nuclear membrane, where it encounters the 18-kD, 5-lipoxygenase-activating protein (FLAP). AA is also translocated to 5-lipoxygenase-activating protein FLAP for presentation to 5-lipoxygenase (5-LO). In addition, upon cell stimulation, cPLA2 is activated and also associates with the nuclear membrane, close to FLAP. The ability of macrophages and dendritic cells to respond appropriately during innate immune responses is likely regulated by 5-LO and 12-LO.52,53 The unstable HPETE is the initial metabolite of each lipoxygenase pathway. HPETE is reduced to the more stable HETE or is converted by 5-LO to LTA4. LTA4 can be converted to LTB4 (in neutrophils and macrophages) or conjugated with reduced glutathione to form LTC4 (in eosinophils, mast cells, endothelial cells, and macrophages). In contrast to lipoxygenase, which is mainly distributed in myeloid cells, LTA4 hydrolase (5,12-dihydroxy-eicosatetraenoic acid), a zinc-requiring enzyme that converts LTA4 to LTB4, is widely distributed. From the cDNA sequence, it was suggested that mRNA for LTA4 may have a short half-life, which could account for the properties of extremely rapid production and shutdown of LTB4 and other eicosanoid biosynthesis.
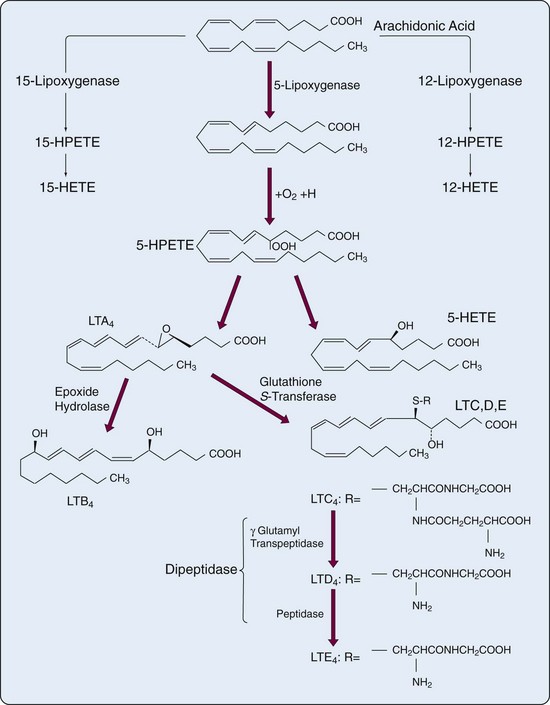
LTA4 can be exported from the cell of origin and converted in other cells by LTA4 hydrolase to LTB4. This variation on the endoperoxide steal—perhaps better called transcellular metabolism—also applies to conversion of LTA4 to LTC4 by LTC4 synthase, a glutathione-S-transferase.54 Although human endothelial cells do not produce terminal products of the 5-lipoxygenase system, they do generate LTC4 from LTA4 provided by neutrophils. LTC4 and its products, LTD4 and LTE4, constitute the biologic mixture previously known as slow-reacting substance of anaphylaxis. LTD4 and LTE4 arise from LTC4 after sequential removal of γ-glutamic acid and glycine from LTC4. The enzyme γ-glutamyl transpeptidase is present in many cells as part of a complex enzymatic system involved in glutathione biosynthesis and amino acid transport. In many systems the major sulfidopeptide LT has been reported to be LTD4, rather than the precursor LTC4. Removal of glycine from LTD4 results in LTE4 with concomitant loss of a significant amount of biologic activity. The principal route of inactivation of LTB4 is by omega oxidation.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


