17 Platelets
Platelets are small circulating cytoplasmic fragments that play a crucial role in hemostasis.
The development of antiplatelet agents offers the promise of new therapeutic modalities.
Platelets are small circulating cytoplasmic fragments that play a crucial role in hemostasis. They are produced in the bone marrow by megakaryocytes. Single platelets circulate freely in the bloodstream; after vascular injury, platelets adhere to the subendothelium, resulting in responses that contribute to formation of the hemostatic plug. These responses include aggregation, secretion of bioactive compounds, and production of procoagulant activity. Platelets also secrete soluble factors that contribute to wound repair by altering vascular tone and permeability, promoting cell growth, and stimulating scavenger cells such as monocytes. During the inflammatory response, many of the activities that lead to hemostasis contribute to inflammation.1 Inflammation initiates clotting, decreasing the activity of natural anticoagulant mechanisms and impairing the fibrinolytic system. Conversely, activated platelets release chemotactic factors that promote leukocyte adhesion, which facilitates their extravasation into inflammatory foci. Platelets secrete a variety of factors that can alter vascular tone and permeability. Last, platelets are a major source of transforming growth factor (TGF)-β, a potent stimulus of fibrosis. Taken together, these activities make platelets contributors to the inflammatory response and to the pathogenesis of systemic rheumatic diseases.2
General Characteristics of Platelets
The plasma membrane of platelets is a typical lipid bilayer, having an extensive series of complex invaginations termed the canalicular system. The role of this surface-connected tubular system seems to be to facilitate the quick release of secreted substances to the extracellular environment. The platelet membrane bears numerous glycoprotein (GP) receptors. Platelet surface phospholipids play an important role in coagulation3 and are a source of arachidonic acid, a precursor of important vasoactive substances such as thromboxane A2, a potent vasoconstrictor and platelet-aggregating agent, and of leukotrienes, which can amplify the inflammatory response. Platelet surface GPs are receptors that mediate adhesion to subendothelial tissue and subsequent aggregation to form the hemostatic plug.4 The largest GP is termed I and the smallest IX. The labels a and b distinguish between two separate electrophoretic bands that initially were considered one (e.g., GPI became GPIa and GPIb). The platelet GPIb-IX-V is an important receptor that binds to von Willebrand factor (vWF) exposed in the subendothelial matrix, causing the attachment of platelets.5 Deficiency of any component of the GPIb-IX-V complex or of vWF leads to the congenital bleeding disorders Bernard-Soulier disease (GPIb-IX-V complex)6 or von Willebrand disease (vWD),7 respectively. vWF, in addition to its important role in hemostasis, has been suggested to promote inflammation, facilitating neutrophil diapedesis by destabilization of the endothelial barrier.8
ADAMTS-13 (a disintegrin-like metalloprotease with thrombospondin type I repeats 13) is a plasma protease that cleaves vWF into smaller multimers, reducing its hemostatic potency.9 Mutations in the ADAMTS-13 gene10 and autoantibodies against ADAMTS-1311 have been shown to cause familial and acquired thrombotic thrombocytopenic purpura, respectively. In mice, ADAMTS-13 has powerful natural antithrombotic activity, and recombinant ADAMTS-13 has proved useful in preventing ischemic brain injury in experimental stroke.12 It has been suggested that ADAMTS-13 might act as a link between thrombosis and inflammation. In inflammatory models, ADAMTS-13 has played an important role in preventing vWF-induced secretion of Weibel-Palade bodies by endothelial cells and, consequently, reducing the adhesion and extravasation of leukocytes.13 Other interactions that contribute to initial platelet adhesion are mediated by collagen receptors GPIa-IIa (integrin α2β1) and GPVI, which bind to collagen in the subendothelial matrix.14 The most abundant platelet surface receptor, GPIIb-IIIa (integrin αIIbβ3), is activated by adhesion to collagen or vWF or by soluble agonists, such as thrombin. After activation, GPIIb-IIIa binds fibrinogen, leading to platelet aggregation.4 Deficiency of this GP results in Glanzmann’s thrombasthenia, a disorder characterized by petechial bleeding and the absence of platelet aggregation and clot retraction.15
The cytoplasm of platelets is rich in actin and myosin, which provide platelets the ability to change shape and to retract clots. Platelet cytoplasm consists of mitochondria, lysosomes, glycogen stores, and three types of granules that contain numerous biologically active molecules (Table 17-1). These granules are classified according to their ultrastructure, density, and contents as alpha granules, lysosomes, and dense granules. Although most of the contents of these granules are made in megakaryocytes, some are taken up from the plasma by megakaryocytes and platelets.
Table 17-1 Platelet Granule Compounds and Granule Membrane Components with a Role in the Hemostatic/Inflammatory Response
| Platelet Granules | Actions | Contents |
|---|---|---|
| Dense granule | Proaggregating factors | Serotonin, histamine, ADP, ATP, Ca2+, Mg2+ |
| Alpha granule | Adhesive glycoproteins | P-selectin, CD31, GPIIb-IIIa, fibronectin, vitronectin, thrombospondin |
| Growth factors | TGF-β, PDGF, EGF, VEGF | |
| Platelet aggregation and chemotaxis | β-Thromboglobulin, PF4 (CXCL4), CC and CXC chemokines | |
| Hemostasis factors | Fibrinogen, vWF | |
| Lysosome | Tissue destruction | Hydrolases, collagenase, cathepsins D and E |
ADP, adenosine diphosphate; ATP, adenosine triphosphate; EGF, epidermal growth factor; GPIIb-IIIa, glycoprotein IIb-IIIa; PDGF, platelet-derived growth factor; PF4, platelet factor-4; TGF, transforming growth factor; VEGF, vascular endothelial growth factor; vWF, von Willebrand factor.
Modified from Rendu F, Brohard-Bohn B: The platelet release reaction: granules’ constituents, secretion and functions, Platelets 12:261, 2001.
Alpha granules contain numerous proteins and growth factors, such as platelet-derived growth factor (PDGF), TGF-β, platelet factor-4 (also referred to as CXCL4), and vWF, which are synthesized in the megakaryocyte.16 Other proteins, such as fibrinogen, enter the alpha granules from the plasma via GPIIb-IIIa receptor–mediated endocytosis.17 P-selectin (CD62P), an adhesion molecule, also is localized in the membrane of alpha granules18 and redistributes to the cell surface during platelet activation. Platelet P-selectin has been implicated in stabilizing platelet aggregates.19 The best documented high-affinity counterreceptor for P-selectin is P-selectin glycoprotein ligand-1 (PSGL-1), a transmembrane sialomucin found on leukocytes and lymphoid cells,20 through whose interaction platelets participate in the inflammatory response.21
Dense granules contain serotonin, adenosine diphosphate (ADP), adenosine triphosphate, and calcium. The dense granule membrane bodies are made in megakaryocytes, but they do not acquire their content of serotonin and calcium until platelets are released into the circulation.22 Another series of intracellular membrane vesicles serves as a reserve to increase membrane surface area on platelet activation.
As stated previously, platelets are small cytoplasmic fragments derived from megakaryocytes. Although megakaryocytes are rare in the bone marrow (approximately 0.1% of all nucleated cells), they are easily recognized by their giant size (50 to 100 µm diameter) and large, multilobed nucleus. Megakaryocytes have two unique characteristics: (1) They undergo a process known as endomitosis, in which the nucleus accumulates many times the normal number of chromosomes, and (2) they have specialized structures in the cytoplasm that permit fragments to be shed, as platelets, into the bloodsteam.23
With a life span of just about 10 days, every day, about 2 × 1011 platelets are released into the bloodsteam of healthy adults by mature megakaryocytes. This quantity can be increased 10-fold under specific conditions. In humans, as in other species, there is an inverse relationship between platelet count and mean platelet volume.24 This suggests that platelet production by bone marrow megakaryocytes is regulated to maintain a constant total platelet mass. The tendency toward a stable platelet mass explains the wide variation in platelet count in healthy donors (150,000/µL to 450,000/µL). Megakaryocytes normally replace about 10% of the platelet mass daily. In response to the increased need for platelets, megakaryocytes modify their number, size, and ploidy. Changes in free levels of thrombopoietin, the main physiologic regulator of platelet production, are responsible for these morphologic and functional adaptations in megakaryocytes.
Thrombopoietin is an 80- to 90-kD GP produced mainly by the liver and released at a constant rate into the circulation. Thrombopoietin acts through its receptor, also known as c-Mpl, which is present in platelets, megakaryocytes, and, to a lesser extent, most other hematopoietic precursor cells. Thrombopoietin prevents apoptosis of megakaryocytes, while increasing their number, size, and maturation,25 but it does not seem to increase the rate of shedding of platelets into the circulation.26 On circulating platelets, thrombopoietin is not a sufficiently strong stimulus to trigger platelet function, but it reduces the threshold for activation by other agonists, such as ADP.27 Binding to the platelet thrombopoietin receptor is the major route of catabolism, however, of circulating thrombopoietin. When the platelet production rate decreases, the platelet mass and the quantity of thrombopoietin receptor decrease; consequently, thrombopoietin concentrations increase and megakaryocyte growth is stimulated. In conditions of high platelet mass (e.g., hypertransfusion of platelets), the number of thrombopoietin receptors increases, thrombopoietin concentrations decrease, and megakaryocyte growth decreases. In addition to thrombopoietin, other soluble factors, such as interleukin (IL)-3, IL-6, IL-11, stem cell factor, or granulocyte-macrophage colony-stimulating factor, seem to promote megakaryocyte growth and maturation. Some of these soluble proteins may play a relevant role in thrombocytosis conditions.28
Hemostasis
When a blood vessel is injured, a complex process involving biochemical reactions and cell-cell and cell-matrix interactions, termed hemostasis, occurs. The initial hemostatic response is mediated by platelets that form the platelet plug (Figure 17-1).
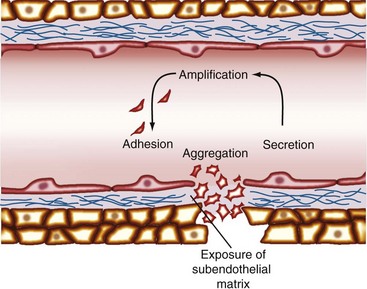
Under physiologic conditions, the undamaged endothelium prevents the adherence of platelets by several mechanisms. These mechanisms include a cell-associated ecto-ADPase (CD39) and the production of nitric oxide and prostacyclin.29 When blood vessel integrity is disrupted, the first reaction is vasoconstriction, which reduces blood loss. Simultaneously, subendothelial matrix elements are exposed, and platelets are rapidly transformed into sticky cellular elements capable of adhering to the underlying surface. Platelet adhesion is initially mediated by the interaction of the GPIb-IX-V receptor complex with vWF in the subendothelial matrix.5 This interaction transduces signals through the GPIb-IX-V complex that activate platelet integrins.30 The activation of GPIa-IIa and GPIIb-IIIa integrins allows the binding to collagen (GPIa-IIa) and vWF (GPIIb-IIIa), mediating the stable adhesion of platelets to the subendothelial surface. In addition to vWF, the active form of GPIIb-IIIa binds fibrinogen.31 The association of soluble fibrinogen with GPIIb-IIIa creates bridges between platelets that result in platelet aggregation and thrombus growth. In concert with aggregation, platelets release their intracellular granules, amplifying the hemostatic response (see Table 17-1).32,33 The outcome is the formation of a platelet plug and triggering of the coagulation cascade, which leads to thrombin generation and resulting fibrin clot formation (Figure 17-2).
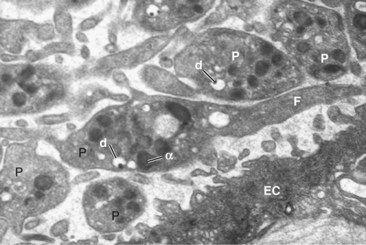
Figure 17-2 Anatomy of a platelet plug. Electron micrograph of a group of platelets (P) attached to an endothelial cell (EC)120 in the initial platelet plug formation. Several dense granules (d) and alpha granules (α) are visible. The central platelet shows long dendritic extensions or filopodia (F).
(Courtesy Dr. Lucio Díaz-Flores.)
One response of platelets to activation by stimuli such as shear stress or collagen is the release of vesicles called platelet microparticles, fragments 0.1 to 0.2 µm in diameter that carry antigens present in intact platelets. These platelet-derived microparticles may play a role in normal hemostasis.34,35 The number of clinical disorders associated with elevated platelet microparticles is increasing,36,37 including several rheumatic diseases in which the number of circulating platelet microparticles seems to be associated with disease activity.38–41 The relevance of platelet-derived microparticles in the physiopathology of those disorders needs to be fully clarified; however, it has been demonstrated that platelets intensify the inflammatory response in joint42 (Figure 17-3).
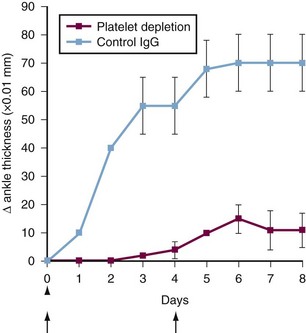
Figure 17-3 Platelets can be involved in the development of arthritis. The passive K/BxN model of arthritis is induced by administration of arthritogenic serum containing antibodies to glucose-6-phosphate isomerase (GPI). The graph shows arthritis severity after K/BxN serum transfer in mice administered a platelet-depleting antibody (red squares) or isotype control (blue squares). Data show the mean ± standard error of the mean (SEM).42 Arrows indicate parenteral administration of platelet-depleting antibody; arrowhead, K/BxN serum administration. These findings suggest that platelets are required for arthritis development in vivo in this model.
Glycoprotein IIb-IIIa
GPIIb-IIIa is a member of a family of cell-adhesion receptors termed integrins. It also is referred to as integrin αIIbβ3 or CD41/CD61. Although integrins are expressed on virtually all nucleated cells, GPIIb-IIIa is restricted to megakaryocytes and platelets. It is the most abundant receptor on the platelet surface, averaging 80,000 copies per platelet. GPIIb-IIIa recognizes at least five different adhesive ligands43: fibronectin, fibrinogen, vWF, thrombospondin, and vitronectin. Cells can modify integrin functions through dynamic modulation of receptor affinity.43 On resting platelets, GPIIb-IIIa does not bind soluble fibrinogen. After platelet stimulation (e.g., by thrombin, collagen, or ADP), GPIIb-IIIa undergoes a conformational change, however, and is converted from a low-affinity to a high-affinity fibrinogen receptor, a process known as inside-out signaling. In this situation, fibrinogen bridges the activated platelets, and platelet aggregation occurs. Simultaneously, the cytosolic portion of the activated GPIIb-IIIa binds to platelet cytoskeleton proteins and mediates platelet spreading and clot retraction in what is referred to as outside-in integrin signaling. GPIIb-IIIa integrates receptor-ligand interactions on the external face of the membrane with cytosolic events in a bidirectional fashion.4 This is the final common pathway for platelet aggregation, regardless of the mode of platelet stimulation. The importance of GPIIb-IIIa integrin is illustrated by Glanzmann’s thrombasthenia, a bleeding disorder caused by mutations in the gene for the αIIb- or the β3-subunit,15 and by the clinical utility of GPIIb-IIIa antagonists as antithrombotic agents in the treatment of thrombotic diseases. Glycoprotein IIb-IIIa inhibitors are now recommended by international guidelines in patients with acute coronary syndromes undergoing percutaneous coronary intervention.
Role of Platelets in the Inflammatory Response
The accumulation of leukocytes in tissue is an essential event for the inflammatory response. The current paradigm of leukocyte extravasation requires a multistep cascade of sequential leukocyte–endothelial cell interactions, in which members of three different families of adhesion receptors participate: selectins, integrins, and the immunoglobulin superfamily.44 Platelets contribute in many ways to leukocyte accumulation in the inflammatory foci (Table 17-2).
Table 17-2 Platelet Components Implicated in the Inflammatory Response
| Platelet Component | Actions | |
|---|---|---|
| Surface molecules | P-selectin (CD62P), PECAM (CD31), GPIbα | Adhesive targets for leukocytes |
| PAF, ROS | Neutrophil activation | |
| CD154 (CD40 ligand) | Agonist for endothelial cells | |
| Soluble factors | Serotonin, histamine | Regulators of vascular permeability |
| β-Thromboglobulin, PF4 | Chemotaxis | |
| Acid hydrolases, ROS | Tissue destruction | |
| PDGF, TGF-β | Cellular mitogens, chemoattractant | |
| End products of platelet procoagulant activity | Thrombin, fibrin | Promote leukocyte accumulation |
GPI, glycosyl phosphatidylinositol; PAF, platelet-activating factor; PDGF, platelet-derived growth factor; PECAM, platelet–endothelial cell adhesion molecule; PF4, platelet factor-4; ROS, reactive oxygen species; TGF, transforming growth factor.
In flowing blood, leukocytes roll on adherent activated platelets, mainly through the interaction of platelet P-selectin with its major leukocyte ligand, PSGL-1.45 This initial rolling of leukocytes on platelet P-selectin is followed by their firm adhesion and subsequent migration—processes that depend on the leukocyte integrin Mac-1 (αMβ2, CD11b/CD18).45,46 Mac-1 adheres firmly to platelets through direct binding to glycoprotein Ibα (GPIbα, CD42b).47 These interactions provide molecular mechanisms for leukocyte recruitment to hemostatic plugs where platelets have been previously deposited in response to vascular injury.48 Parallel lines of investigation have shown that resting platelets are able to roll on activated endothelial cells, apparently through an interaction between PSGL-1 expressed in platelets and the endothelial P-selectin.49 The physiologic function of platelet rolling on stimulated endothelial cells needs to be clarified. If this contact results in activation of platelets, however, those platelets may release proinflammatory mediators, such as cytokines, chemokines,50,51 and eicosanoid precursors,52 or growth factors that stimulate tissue healing. Activated platelets in circulation stimulate secretion of Weibel-Palade bodies from endothelial cells in vivo; this leads to P-selectin–mediated leukocyte rolling.53 Given the important role of platelet P-selectin in chronic inflammatory processes,54,55 this effect of activated platelets might represent an important pathway of platelet-induced inflammation.
In addition to the adhesion molecules, activated platelets express on their surface two major proinflammatory mediators: platelet-activating factor (PAF) and CD40 ligand (CD154). PAF is a potent platelet-aggregating phospholipid produced by macrophages, mast cells, platelets, endothelial cells, neutrophils, and monocytes. Upon cell activation, PAF is rapidly synthesized and translocated to the plasma membrane of endothelial cells, where it recognizes its receptor in neutrophils, resulting in β2 integrin–mediated adhesion of leukocytes to the endothelial surface.56 In the same way, PAF can signal neutrophils when it is displayed on the surface of adherent activated platelets acting in cooperation with P-selectin to tether neutrophils.56 The biologic action of PAF is physiologically inactivated by plasma and cellular acetylhydrolase.57 A role of PAF in the pathogenesis of chronic inflammatory arthritis has been proposed58; however, a well-controlled clinical trial failed to show any beneficial effect of a PAF antagonist in patients with active rheumatoid arthritis (RA).59
CD40 is a transmembrane protein member of the tumor necrosis factor (TNF) receptor family. CD40 is present on many cells, including B cells, monocytes, macrophages, dendritic cells, and vascular endothelial cells. Platelets are the major peripheral blood source of CD154, the ligand of CD40, and they express it on their surface within seconds of exposure to an agonist. The interaction of CD154 on activated platelets with CD40 on endothelial cells causes a proinflammatory reaction of the endothelium characterized by expression of inflammatory adhesion molecules, such as E-selectin, vascular cell adhesion molecule-1 (CD106), and intercellular adhesion molecule-1 (CD54), and secretion of the chemokines IL-8 (CXCL8) and monocyte chemotactic protein-1 (CCL2).60 CD154 expressed on activated platelets can provide a potent stimulus to the inflammatory response. Clinical data from an open-label study suggested that the blockade of CD154 with a biologic may induce a prothrombotic state in patients with lupus nephritis through a mechanism not clarified.61 A phase II, double-blind, placebo-controlled study evaluating the safety and efficacy of a humanized monoclonal antibody against CD154 in patients with active systemic lupus erythematosus failed to show clinical efficacy. In this study, the type and frequency of adverse events were similar between the intervention and placebo groups.62
When platelets adhere, they release numerous growth factors, such as PDGF, TGF-β, and other factors that are chemotactic for monocytes, macrophages, and fibroblasts. These growth factors may play an important role in the chronic inflammatory response by mediating a fibroproliferative response. PDGF is a homodimer or heterodimer molecule of A and B chains63 produced by platelets, monocytes or macrophages, endothelial cells, and vascular smooth muscle cells (under some conditions). This molecule plays an essential role in tissue repair and wound healing.64 PDGF is a potent mitogen and chemoattractant for smooth muscle cells, connective tissue cells, and macrophages65–68; it contributes to the formation of lesions of atherosclerosis,68,69 a disorder strongly related to the inflammatory response.70 It has been shown that PDGF is a potent mitogen for synovial fibroblasts isolated from patients with RA.71
TGF-β has three isoforms (TGF-β1, TGF-β2, and TGF-β3) secreted by virtually all cell types as latent complexes that need to be processed to exhibit biologic activity.72 Several effects have been associated with TGF-β: (1) It is chemotactic for various cell types, including leukocytes; (2) it inhibits proliferation of most cells; (3) it induces the synthesis and deposition of extracellular matrix; and (4) it stimulates the formation of granulation tissue.73 The net result is that TGF-β is mainly an inhibitor of the inflammatory response.74 Carefully regulated expression of active TGF-β is essential for resolution of inflammation and repair. Systemic administration of TGF-β1 has antagonized the development of polyarthritis in susceptible rats.75 Overproduction of this cytokine has been associated with several fibrotic processes.76,77 TGF-β is a major cytokine involved in the pathogenesis of fibrosis in systemic sclerosis.78 Blockade of cell surface molecules capable of activating latent TGF-β and blockade of ligand by antibody, soluble TGF-β receptors, and a recombinant latency-associated peptide, as well as inhibitors for ALK5 and Smad3, are potential strategies for abolishing the pathologic activation of TGF-β in systemic sclerosis.
Several reactive oxygen species are released from unstimulated platelets and after platelet stimulation with agonists such as collagen or thrombin.79,80 Because reactive oxygen species have been implicated in direct tissue injury and in inflammatory reactions through promotion of adhesive interactions between inflammatory and endothelial cells,81 reactive oxygen species originating from platelets may act as an autocrine or paracrine mediator that participates in the amplification of the inflammatory response in disorders such as rheumatic diseases.









