74 Pathogenesis of Ankylosing Spondylitis and Reactive Arthritis
Genetics plays a major role in the etiology of ankylosing spondylitis.
The gene with the greatest contribution to ankylosing spondylitis is HLA-B27.
A major mediator of inflammation is tumor necrosis factor (TNF).
Bone destruction occurs along with pathologic new bone formation in ankylosing spondylitis.
Reactive arthritis is initiated by infection outside the joints.
Ankylosing spondylitis and reactive belong to the family of spondyloarthritis. Reactive arthritis (ReA), is clearly distinct in that it is induced by an episode of acute infection. Accordingly, the pathogenesis of these two diseases will be described in separate sections. For ankylosing spondylitis, it is commonly recognized that two major but “uncoupled” processes coexist inflammation and abnormal bone formation.1 The process of bone formation in this disease has become a major research frontier.
Pathogenesis of Ankylosing Spondylitis
The causes of ankylosing spondylitis are multifactorial and involves a number of interlinking pathways. A considerable number of critical factors have been identified. They can be classified into two groups. One group consists of mediators that are midstream in the pathways leading to inflammation. These mediators are targets of current therapies. The second group of critical factors contributing to ankylosing spondylitis is genetics. How should the degree of contribution of each factor be estimated? For therapeutics, the usefulness of each therapy will be ranked by a statistical value known as effect size. In pharmacology, effect size reflects the size of the difference in outcome between groups receiving a drug compared with a group receiving placebo, taking into account the degree of variability within groups. The higher the effect size, the more effective is the therapy. For ranking the genes, we will use the statistical value known as the population attributable risk. For each gene, this value represents the incidence of disease in a population that would be eliminated if the gene is absent. The higher the population attributable risk, the greater the contribution to the disease.2
Causes of Ankylosing Spondylitis That Are Targets of Therapies
It has been known for more than three decades that most patients with ankylosing spondylitis demonstrate a highly satisfactory response to treatment with nonsteroidal anti-inflammatory drugs (NSAIDs), including the cyclooxygenase-2 (COX-2) inhibitors (coxibs).3 The effect size for spinal pain is 1.11, which means that there is a 77% probability that a randomly selected NSAID-treated patient will show a better response than a randomly selected placebo-treated patient.4 This effect size is much greater than in patients with nonspecific chronic low back pain.5 Because the molecular targets of most NSAIDs are COX-1 and COX-2, and the target of the coxibs is restricted to COX-2, it can be concluded that one of the pathways causing spinal pain in ankylosing spondylitis is the COX-2 pathway.6 Indeed, the COX-2 pathway generates prostaglandins and thromboxanes, which are strongly proinflammatory. The response of spinal pain in ankylosing spondylitis to NSAIDs is indirect evidence that inflammation is responsible for the pain. In fact, the spinal pain of ankylosing spondylitis is designated as being “inflammatory” rather than “mechanical.” The presence of spinal inflammation is validated by magnetic resonance imaging (MRI) observations.7 Although we know that the COX-2 pathway is responsible in part for the symptoms of ankylosing spondylitis, no information is available as to what triggers this pathway or which downstream mediators are most important.
When responses to NSAIDs are not satisfactory, some patients will be given a trial of a disease-modifying antirheumatic drug (DMARD). Most of the conventional DMARDs useful for rheumatoid arthritis such as methotrexate are much less effective in ankylosing spondylitis.4 Similar to rheumatoid arthritis, for patients who are resistant to treatment with NSAIDs or conventional DMARDs, there is a very high rate of clinical response to biologically generated agents that target tumor necrosis factor (TNF). Currently, several of these biologics have been approved for the treatment of ankylosing spondylitis in the United States. All are very effective in controlling symptoms. The effect size for etanercept, for example, is 2.25, which means that more than 90% of randomly selected patients will respond better than another randomly selected patient who is treated with placebo.4 Hence there is little doubt that TNF is a major player in causing the symptoms of ankylosing spondylitis. TNF is a cytokine generated via innate and adaptive immunities by several types of cells, including macrophages, T lymphocytes, and mast cells. TNF is not a factor with a single pathway inducing a single event in a single type of cell. Rather, it is pleiotropic in affecting many cell types, and it induces a network of cytokines and chemokines and other mediators of inflammation.8 Although it is certain that TNF is a major early upstream mediator in ankylosing spondylitis, no agreement has been reached as to which cell types are specifically responsible for generating TNF, or which processes are disease-specific targets of TNF.
Assessment of Degree of Contribution by Environmental Versus Genetic Factors and Identification of Genetic Factors
Degrees of contribution of environmental versus genetic factors have been estimated from the degree of concordance among twins. According to those analyses, genetics contributes to more than 90% of the total cause of ankylosing spondylitis. In addition, if there are environmental causes, they are probably ubiquitous, such as enteric bacteria.9 Indeed, rats carrying the human ankylosing spondylitis–causing transgene HLA-B27 do not develop arthritis when bred in a germ-free environment. However, arthritis will develop once they are transferred to a regular environment.10 It is thought that the development of arthritis in these rats is related to commensal bacteria, especially in the gastrointestinal tract. In human ankylosing spondylitis, the only enteric bacterium that has been incriminated is Klebsiella pneumoniae. As a group, patients with ankylosing spondylitis have higher mean antibody titers to Klebsiella compared with control subjects.11 However, evidence supporting Klebsiella as a cause of ankylosing spondylitis is far less strong than that which supports, for example, that streptococcal infections are a cause of rheumatic fever.
Over the past decade, most of the researchers studying the pathogenesis of ankylosing spondylitis have focused on identification of arthritis-causing genes. According to statistical modeling based on family studies, the disease is caused by up to nine genes with multiplicative interactions among loci.12 The most important gene, HLA-B27, was discovered in 1973.13,14 It contributes about 30% of the heritability of the disease. About three decades later, with the development of the technique of genome-wide association study of nonsynonymous single-nucleotide polymorphisms (SNPs), at least four other loci encoding known structural gene sequences have now been reported. In the Wellcome Trust Case Control Consortium and the Australo-Anglo-American Spondylitis Consortium studies, when these genes are ranked according to their degree of contribution, they are listed as follows: HLA-B27, ERAP1 (endoplasmic reticulum amino peptidase 1, previously known as aminopeptidase-regulating tumor necrosis factor receptor shedding 1, abbreviated as ARTS-1), IL-23R (interleukin 23 receptor), IL-1R2 (interleukin 1 receptor, type II), and ANTXR2 (anthrax toxin receptor, also known as capillary morphogenesis protein 2, or CMG2). The population attributable risks for the first three are 90%, 26%, and 9%, respectively. Those for IL-1R2 and ANTXR2 are probably less than 5%.15,16 Hence, HLA-B27 is the largest contributor.
How HLA-B27 Induces Ankylosing Spondylitis
The most important gene in ankylosing spondylitis is HLA-B27. In most populations, it is present in more than 90% of patients and in less than 10% of the general population.17 Rats carrying the HLA-B27 transgene develop arthritis and colitis.18,19 Using gene terminology from microbiology, it is described as being “essential” for ankylosing spondylitis. Because the association between HLA-B27 and ankylosing spondylitis has been known for more than three decades, the search for how HLA-B27 causes ankylosing spondylitis has been the Holy Grail of many scientists, and an enormous number of publications have been generated. HLA-B27 is an allele of the HLA-B locus of the human leukocyte antigen (HLA) class I antigens. Multiple subtypes of HLA-B27 have been defined.20 Most experiments attempting to identify the mechanisms of how HLA-B27 mediates arthritis have been carried out with the more common B27*05 and B27*04 subtypes. Multiple hypotheses have been reported regarding how HLA-B27 molecules mediate arthritis. At least three hypotheses are currently under active investigation.
Arthritogenic Peptide Hypothesis
The arthritogenic peptide hypothesis is based on the classic structure and the canonical function of HLA-B alleles. As in all HLA class I molecules, a classic HLA-B27 molecule is a trimolecular complex of a polymorphic HLA class I heavy chain together with a monomorphic light chain (β2-microglobulin [β2m]) and a single highly variable peptide (Figure 74-1). Most of the peptides are nanomers that conform to a strict motif, usually with arginine as the second amino acid. The motif of peptides that bind to HLA-B27 is different from that of peptides that bind to other HLA-B alleles or to HLA-A or -C molecules.21,22 These polymorphisms probably are driven by evolution to survey against ever changing pathogens such as the influenza viruses. The canonical function of HLA-A and -B molecules is to present peptides derived from such intracellular pathogens to CD8+ T lymphocytes to generate a protective adaptive immune response.23 The reason why this is vulnerable to autoimmunity is that most of the peptides complexed with HLA-A and -B molecules are derived from self-proteins. In health, the hosts are tolerant to all these self-peptides.
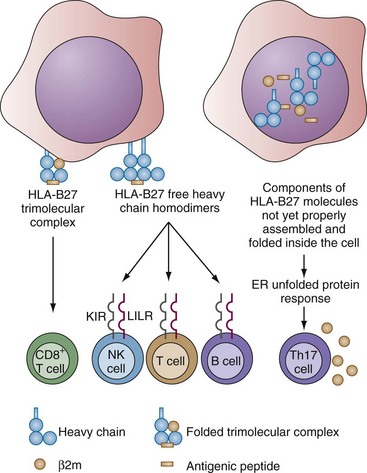
The arthritogenic peptide hypothesis postulates that in the case of ankylosing spondylitis, there is a breakdown of tolerance to certain self-peptides, and this breakdown is a consequence of mimicry between the self-peptides and certain pathogen-derived and arthritis-causing peptides.24 Identification of these arthritogenic peptides demands a great deal of fundamental information concerning the quaternary structures of the HLA-peptide complexes, as well as the dynamics of these structures, the peptide repertoires of candidate pathogens, the repertoires of self-peptides, and last, the repertoires of T cell receptors. At this point, enormous detail has been generated concerning the structures of HLA-peptide complexes. Reactivity against several self-peptides has also been reported. Many yet untested candidate peptides have recently been identified and will require testing in the future, perhaps with newly developed immunoproteomic techniques.25,26
Free Heavy Chain Hypothesis
The free heavy chain hypothesis is based on the observation that HLA-B27 molecules can exist on the cell surface as free heavy chains free of the more usual association with β2m or peptides with the classic HLA-B27 motif (see Figure 74-1). These free heavy chains exist as stable dimers and are capable of engaging allele-specific receptors on natural killer (NK) cells and T lymphocytes. Receptors for HLA-B27 free heavy chain dimers are the KIR3DL1*001 allele of KIRs (a family of immunoglobulin-like receptors on NK cells and certain subsets of T cells) and the LILRA1 and LILRB2 alleles of LILRs (a family of leukocyte immunoglobulin-like receptors on NK cells, T and B cells, and cells of the myeloid lineage).27 The free heavy chain hypothesis postulates that engagement of these receptors will generate arthritis-causing events. This hypothesis is supported by actual observations of ankylosing spondylitis patients. Such HLA-B27 heavy chain dimers are found on the cell surfaces of mononuclear cells, along with expansion of KIR3DL2-expressing NK and CD4+ T cells responding to such dimers.28,29
Unfolded Protein Hypothesis
The unfolded protein hypothesis is different from the previous two in that it is concerned not with activities of HLA-B27 on the cell surface, but with activities inside the cells (see Figure 74-1). Like most surface proteins, the HLA-B27 heavy chains are synthesized linearly into a processing organelle inside the cell by the endoplasmic reticulum (ER). At first, these newly synthesized proteins do not have any conformation and are described as being “unfolded.” Then, they are stepwise driven into a series of conformations through sequential complexing with a corresponding series of ER chaperones. These ER chaperones generate conformations in their target proteins by binding to their inappropriately exposed hydrophobic domains, as well as to underglycosylated residues. Through a series of cycles of binding and release with a series of ER chaperones, formation of disulfide bonds, and pairing with the β2m and a peptide, an HLA-B27 molecule will mature into a quaternary and stable form, which then will be transported to the cell surface.30
This sequence of events takes place over a period of time. Compared with a few other common HLA alleles, HLA-B27 heavy chains have more prolonged retention times inside the ER. In addition, with HLA-B27, more partially misfolded and unfolded forms are found inside the ER compared with other HLA alleles. Reasons include aberrant disulfide bond formation and multimer formation among the heavy chains, among others. These partially folded or misfolded HLA-B27 proteins remain for a time sequestrated inside the ER, being complexed with chaperones such as the “immunoglobulin heavy chain–binding protein,” abbreviated as BiP (GRP78, 78-kD glucose-regulated protein). BiP is a sensor for accumulation of misfolded proteins. It initiates several ER processes, which together are termed unfolded protein response (UPR).31,32 UPR can be cytoprotective or might lead to apoptosis. UPR is associated with a number of diseases, including cancer, diabetes, atherosclerosis, and neurologic disorders. For ankylosing spondylitis, it has been observed in an in vitro system that UPR with HLA-B27 polarizes the cells in response to lipopolysaccharide to cross-talk with the cytokine systems by generating interleukin (IL)-23. The HLA-B27 unfolded protein hypothesis postulates that HLA-B27 induces an unfolded protein response, which, in conjunction with activation by pattern recognition receptors (PRRs) such as those for lipopolysaccharide, would generate proinflammatory cytokines to such a degree as to cause arthritis.32,33

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


