68 Nutrition and Rheumatic Diseases
Nutritional factors can have proinflammatory or anti-inflammatory effects, or both.
Many patients with rheumatic disease believe diet plays an important role in their symptoms.
Nutrition plays a role in the management of most chronic diseases. Physicians provide dietary advice to patients with diabetes, heart disease, and obesity as part of standard clinical care. Although the role of diet and nutrition is well established in the etiology and management of gout, the role of nutrition in other rheumatic diseases such as rheumatoid arthritis (RA) is less well accepted. In general, dietary advice is not part of standard clinical practice for patients with inflammatory rheumatic diseases. Despite a widespread lack of conviction among physicians about the role of nutrition, many people with arthritis believe food plays an important role in their symptom severity and approximately 50% will have tried dietary manipulation in an attempt to improve their symptoms.1
Nutrition and the Inflammatory Process
Key Points
Omega-3 fatty acids are immunoregulatory.
Vitamin D has multiple immunosuppressive effects.
Antioxidants can be acquired through the diet.
Adipose tissue is metabolically active and has effects on the inflammatory response.
Role of Omega-3 Fatty Acids and the Inflammatory Process
Fatty Acid Biochemistry
The critical process linking fatty acids and inflammation is the metabolism of AA and EPA to eicosanoids, which act as inflammatory mediators. AA is metabolized via cyclooxygenase (COX) to n-6 eicosanoids (prostaglandin [PG] E2, thromboxane [TX] A2 or via 5-lipoxygenase [5-LOX] to n-6 leukotrienes [LTs]). In comparison, EPA is metabolized via COX and 5-LOX to n-3 PGs and LTs, respectively. In comparison to the n-6 eicosanoids produced from AA, EPA is a poor COX substrate such that n-3 PGs are not as readily produced (Figure 68-1). EPA and DHA competitively inhibit production of most n-6 eicosanoids, with prostacyclin (PGI2) being an exception. Increased dietary consumption of n-3 fatty acids such as EPA increases the proportion of EPA incorporated in cellular membranes and tissues partly at the expense of AA incorporation. The net result is an alteration in the balance of n-3/n-6 eicosanoid production (see Figure 68-1).
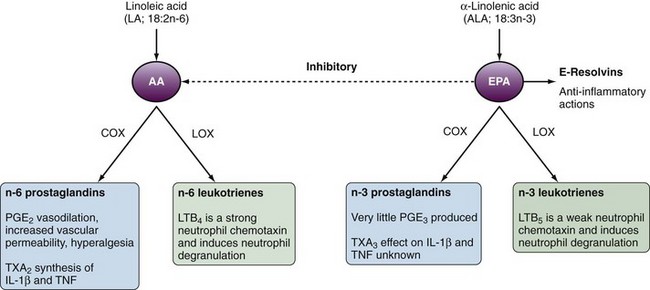
Proinflammatory Actions of Eicosanoids
In general the n-6 eicosanoids (PGE2 and TXA2) are proinflammatory, whereas the n-3 eicosanoids are either less potent in their effects (TXA3) or less abundant (PGE2). TXA2 promotes interleukin-1β (IL-1β) and tumor necrosis factor (TNF) production by mononuclear cells,2 whereas PGE2 results in vasodilatation, increased vascular permeability, and hyperalgesia (see Figure 68-1). PGE3 is edemogenic, although little is produced. LTB5 is 10 to 30 times less potent than LTB4 as a neutrophil chemotaxin.
Effect of n-3 Fatty Acids on Proinflammatory Cytokine Production
IL-1β and TNF production may be reduced as a consequence of dietary n-3 fatty acid supplementation. Although some of this cytokine inhibition is mediated through effects on eicosanoids, there also appears to be eicosanoid-independent cytokine inhibition. For example, fatty acids may have direct effects on intracellular signaling mechanisms including nuclear factor κB (NFκB) and PPAR-γ, thereby affecting cytokine production.3
The resolvins (resolution phase interaction products) are derived from n-3 fatty acids via COX-2 with increased production in the presence of aspirin. Resolvins derived from EPA are known as E-resolvins, whereas those derived from DHA are known as D-resolvins. The resolvins have a variety of anti-inflammatory actions including inhibition of TNF-induced transcription of IL-1β and inhibition of human polymorphonuclear leukocyte transendothelial migration (for review, see Kohil and Levy’s study4). The identification of resolvins provides another mechanism through which n-3 fatty acids contribute to inhibition of inflammatory cytokine production.
Effects of n-3 Fatty Acids on MHC Expression
The number of MHC molecules expressed on antigen-presenting cells (APCs) is an important determinant of T cell response to antigen. Patients with RA have high levels of MHC class II expression on T cells and synovial lining cells.5 In vitro studies show that EPA and/or DHA reduces monocyte expression of HLA-DR and HLA-DP molecules and reduces the ability of monocytes to present antigen to autologous lymphocytes.6 Thus n-3 fatty acids may have an anti-inflammatory effect via suppression of pathogenic T cell activation through inhibition of APC function.
Effect of n-3 Fatty Acids on Adhesion Molecule Expression
Adhesion molecules expressed on endothelial cells and leukocytes mediate the transit of cells from the circulation into tissues. Intercellular adhesion molecule-1 (ICAM-1) and its cognate receptor, leukocyte function-associated antigen (LFA)-1, have been shown to be important in migration of leukocytes into inflamed synovium in animal models.7 ICAM-1 blockade has also been reported to reduce disease activity in RA.8 In vitro n-3 fatty acids decrease human monocyte ICAM-1 and LFA-1 expression.6 In addition, dietary n-3 fatty acid supplementation reduces soluble ICAM-1 and vascular cell adhesion molecule-1 (VCAM-1) plasma concentrations,9 although whether cell surface expression of these adhesion molecules is also reduced has not been reported.
Effect of n-3 Fatty Acids on Degradative Enzymes
Proteinases have a pivotal role in cartilage degradation and bone erosion. n-3 Fatty acids added in vitro can suppress proteinases ADAMTS-4, ADAMTS-5, and MMP-3 in IL-1α stimulated bovine chondrocytes.10 This inhibition of chondrocyte proteases is a mechanism through which n-3 fatty acids may inhibit cartilage degradation and bone erosion.
The RANK/RANKL/OPG pathway is also important in bone pathophysiology in RA. Increased RANK/RANKL and decreased OPG contribute to bone erosion. Three months of dietary fish oil supplementation has been reported to decrease the RANK/OPG ratio, which may help prevent bone resorption that leads to erosions.11
Importance of the Balance of n-3 and n-6 Fatty Acids in the Inflammatory Process
The balance of AA and EPA can be altered through dietary fatty acid intake. In humans, the conversion of dietary ALA to tissue EPA is inefficient and fish/fish oils are a more effective way to increase EPA and DHA in tissues. Changes in AA/EPA ratios in tissues have downstream effects on eicosanoid production and the resulting proinflammatory/anti-inflammatory environment. Dietary supplementation with fish oil in humans results in decreased production of PGE2,12 TXA2,12 and LTB413 with increased production of TXA314 and LTB5.15 These data provide a mechanistic basis for beneficial effects of dietary n-3 fatty acid supplementation in the control of inflammatory diseases. Dietary fish oil supplements have been shown to increase vascular production of prostacyclin (PGI2).16 Although the role of PGI2 in inflammation is not well defined, it is a potent vasodilator and inhibits platelet aggregation, as well as disaggregating platelets. These effects likely contribute to the protective effects of dietary fish and fish oils against thrombotic vascular events. Importantly, patients with several of the major rheumatic diseases (e.g., RA, systemic lupus erythematosus [SLE] and gout) are at high risks for serious cardiovascular events and mortality, to which nonsteroidal anti-inflammatory drug (NSAID)–associated COX-2 inhibition may also contribute.
Vitamin D and the Inflammatory Process
DCs have a central role in activation of the immune system and in response to self. 1,25(OH)2D3 inhibits the differentiation of monocyte precursors into mature DCs, downregulates expression of MHC class II molecules on DCs, inhibits IL-12 production, and promotes DC apoptosis, thereby inhibiting DC-depenent T cell activation.17,18 In addition, 1,25(OH)2D3 can promote DC expression of tolerizing functions, which instruct T regulatory (Treg) cells, which in turn may inhibit the development of autoimmunity.19 Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production including TNF, IL-6, and IL-1α.20 Vitamin D has direct effects on T cells, in particular inhibition of proliferation and cytokine production by Th1 cells, and may enhance Th2 cytokine production.21 1,25(OH)2D3 has also been shown to reduce Th17 cell differentiation through its effects on DCs, as well as direct effects on Th17 cells leading to reduced IL-17A production.19,22 1,25(OH)2D3 inhibits the proliferation of activated B cells, induces activated B cell apoptosis, and inhibits plasma cell differentiation and immunoglobulin secretion.23 Thus vitamin D deficiency may have a role in the etiology of B cell–mediated autoimmune disorders, whereas vitamin D supplementation may have beneficial effects in B cell–mediated autoimmune diseases such as SLE and RA.
Obesity and the Inflammatory Process
Adipose tissue was originally thought to be simply a fat store. However, it is now recognized that adipose tissue and adipocytes are metabolically active and contribute to systemic inflammatory responses (Figure 68-2). Adipocytes release the proinflammatory cytokines TNF, IL-1β, and IL-6. IL-6 enters the systemic circulation and increases C-reactive protein and serum amyloid A production by the liver.
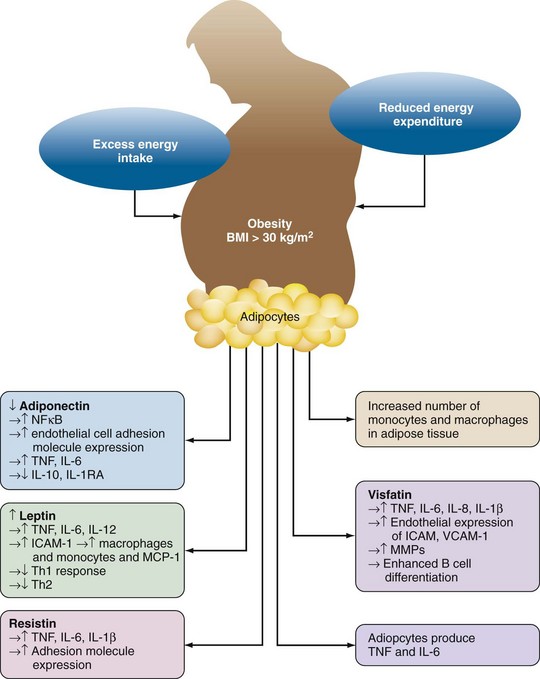
Adipocytes produce adipokines; leptin, resistin, and visfatin (proinflammatory); and adiponectin (anti-inflammatory) (see Figure 68-2). Although the primary function of leptin is appetite control, it also has a number of proinflammatory actions. Leptin increases the expression of adhesion molecules such as ICAM-1 and monocyte chemoattractant protein-1 (MCP-1) thereby favoring recruitment of monocytes/macrophages into adipose tissue. Leptin also increases IL-1β, TNF, and IL-6 production by monocyte/macrophage. Leptin activates T cells, increasing proliferation of Th1 cells while inhibiting Th2 cells. The anti-inflammatory effects of adiponectin include inhibition of TNF-induced adhesion molecule expression; inhibition of NFκB, a pivotal intracellular factor in activation of inflammatory responses; and production of the anti-inflammatory cytokines IL-10 and IL-1RA. Production of adiponectin is inhibited by TNF, which thereby helps sustain the proinflammatory alteration of homeostasis found in obesity. Resistin is produced by mononuclear cells and adipocytes. Resistin increases macrophage/monocyte and adipocyte production of TNF, IL-6, and IL-1β and also increases expression of the adhesion molecules ICAM-1, VCAM-1, and MCP-1.24 Visfatin, which is produced by lymphocytes and adipocytes, has similar proinflammatory effects including induction of IL-8, IL-6, IL-1β, and TNF; increased endothelial expression of ICAM-1, VCAM-1, and MMPs; and enhancement of B cell differentiation.24 The net result of obesity is thus an inflammatory state associated with an increase in circulating C-reactive protein.
Probiotics and the Inflammatory Process
Probiotics may exert their putative anti-inflammatory effects by interacting with intestinal epithelial cells. Resultant modulation of intestinal microflora and fortification of intestinal barrier function may lead to altered exposure of the immune system to microbes and direct effects on immune cells within the intestine. Associated effects relevant to anti-inflammatory responses include accumulation of CD4+ Treg cells in inflamed areas25; inhibition of DC activation26; antagonism of NFκB, which leads to decreased IL-1β and TNF production; and induction of the inflammation-modulating cytokine TGF-β.
Summary
Dietary components can have a variety of effects on the inflammatory process and on bone destructive pathways (Table 68-1). Thus alterations in diet may have effects on both the risk and management of rheumatic diseases, as discussed later.
Table 68-1 Effects of Dietary Components on Inflammatory and Bone Destructive Pathways
| n-3 Fatty Acids |
| Decreased production of n-6 derived eicosanoids (PGE2, TXA2, LTB4), which have proinflammatory effects |
| Increased production of n-3–derived eicosanoids (PGE3, TXA3, LTB5), which in general are less proinflammatory |
| Decreased IL-1β and TNF production |
| Increased production of resolvins |
| Decreased major histocompatibility complex II expression by antigen-presenting cells |
| Decreased adhesion molecule expression: ICAM, VCAM, LFA |
| Decreased expression of matrix metalloproteinases |
| Alteration in RANK/OPG ratio |
| Vitamin D |
| Inhibition of monocyte differentiation into DCs and promotion of DC apoptosis |
| Induction of tolerogenic Tregs with enhanced suppressive activity |
| Inhibition of monocyte/macrophage IL-1β and TNF production |
| Inhibition of Th1 cell proliferation and cytokine production |
| Enhancement of Th2 cytokine production |
| Reduction in Th17 cell differentiation and IL-17A production |
| Inhibition of proliferation of activated B cells, differentiation of activated B cells and immunoglobulin secretion |
| Antioxidants |
| Scavenge reactive oxygen species Reduce proinflammatory eicosanoids, TNF and IL-1β |
| Probiotics |
| Generation of CD4+ Tregs in inflamed areas |
| Inhibition of DC activation |
| Decreased IL-1β and TNF production via inhibition of nuclear factor κB |
| Increased production of transforming growth factor-β |
DC, dendritic cell; ICAM, intercellular adhesion molecule; IL, interleukin; LFA, leukocyte function-associated antigen; LTB, leukotriene B; PGE, prostaglandin E; RANK/OPG, receptor activator of nuclear factor κB/osteoprotegerin; Th, T helper; TNF, tumor necrosis factor; TXA, thromboxane A; VCAM, vascular cell adhesion molecule.
Nutrition in the Etiology of Rheumatic Diseases
Rheumatoid Arthritis
Key Points
Assessing dietary intake is difficult, and identifying a single dietary factor may not be possible.
Omega-3 fatty acids may protect against RA.
The association between vitamin D and RA remains unclear.
Omega-3 Fatty Acid Consumption
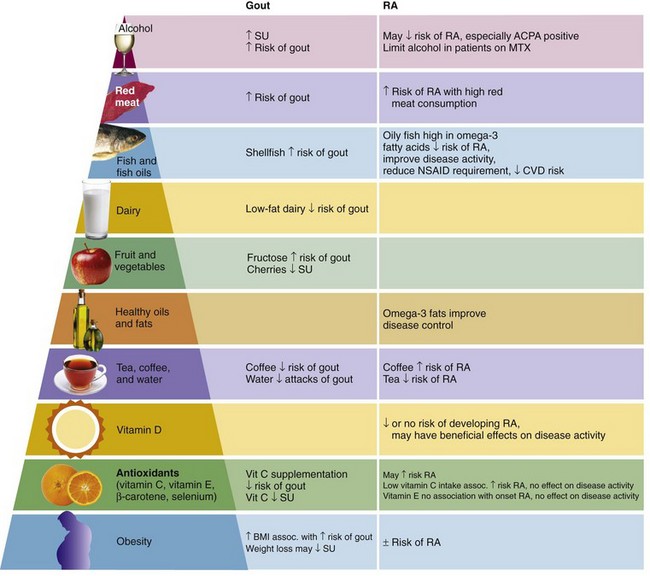
The long chain n-3 fats, EPA and DHA, are most abundant in fish and fish oils. A protective effect against RA of fish intake has been reported. For example, the Seattle Women’s Health Study showed reduced risk for developing RA in subjects consuming two or more fish meals per week with an adjusted odds ratio (OR) of 0.57 (95% confidence interval [CI], 0.35 to 0.93) compared with subjects consuming less than one fish meal per week.27 A more recent population-based, case-controlled study reported a modest decrease in the risk of RA in subjects who consume oily fish one to seven times per week compared with those who seldom or never consumed fish (OR, 0.8; 95% CI, 0.6 to 1.0), which did not change even allowing for RF and anticyclic citrullinated protein (CCP) antibody status.28
Red Meat and Protein Consumption
High consumption of red meat has been associated with an increased risk of inflammatory polyarthritis (OR, 1.9; 95% CI, 0.9 to 4.0).29 Although one study reported that meat and offal were associated with an increased risk of developing RA,30 this was not confirmed in other studies.31,32 Whether the association between red meat consumption and inflammatory arthritis is causative remains unclear, although the presence of significant amounts of AA in red meat may provide some explanation for the association.
Tea and Coffee Consumption
Tea and coffee have been identified as potential risk factors for the development of RA. In the Finnish National Health Study, consumption of four or more cups of coffee per day was associated with an increased risk of RF-positive, but not RF-negative, RA after adjustment for potential confounders such as age, smoking, and gender (relative risk [RR], 2.2; 95% CI 1.13 to 4.27).33 By contrast, the Iowa Women’s Health Study reported no association between daily caffeine intake and risk for RA. However, women who consumed four or more cups of decaffeinated coffee per day were at increased risk for RA compared to non–coffee drinkers (RR, 2.58; 95% CI, 1.63 to 4.06). Furthermore, women who consumed three or more cups of tea per day had a reduced risk of RA (RR, 0.39; 95% CI, 0.16 to 0.97).34 More recent studies have not shown an association between tea/coffee and RA.31,35
Although the epidemiologic evidence for an association between coffee/tea and RA is equivocal, interest has been sustained by the presence of metabolically active agents in these beverages. For example, the major catechin in tea inhibits induction of inducible nitric oxide synthase (iNOS) by stimulated macrophages.36 iNOS generates highly reactive free radical products while di-imidating substrate arginine moieties to citrulline. Citrullinated peptides/proteins are recognized immunogens that provide a focus for autoimmunity in RA.
Alcohol Consumption
Alcohol consumption may reduce the risk of developing RA. In a case-control study of 515 patients with RA, alcohol consumption was associated with a reduced risk of anti-CCP positive RA.37 A dose-dependent inverse relationship between alcohol consumption and risk of RA has also been demonstrated from two independent case-control studies (Swedish EIRA and Danish CACORA).38 The reduction in risk of RA was more pronounced in patients with the shared epitope compared those without the shared epitope and most pronounced in smokers with the shared epitope.38
With regard to candidate mechanisms for this putative reduction in risk for RA, alcohol has been shown to downregulate the production of proinflammatory cytokines and upregulate production of the anti-inflammatory cytokine IL-10.39,40 Furthermore, in a murine model of arthritis, ethanol almost totally prevented the development of collagen-induced arthritis and in those mice that did develop arthritis, disease was less severe. These anti-inflammatory effects of ethanol were associated with reduced leukocyte migration, downregulation of NFκB, and reduced production of the proinflammatory cytokines IL-6 and TNF but not the anti-inflammatory cytokine IL-10.41
Vitamin D
As noted earlier, vitamin D has anti-inflammatory actions. Although vitamin D has been implicated in reducing the risk of the autoimmune diseases, diabetes,42 and multiple sclerosis, the association with risk of RA is less clear.43
The Iowa Women’s Health Study reported that a higher intake of vitamin D was associated with a reduced risk of RA (RR, 0.67; 95% CI, 0.44 to -1.00, P = 0.05) in women aged 55 to 69 years.44 However, in a more recent large study of 186,389 women followed for 22 years, there was no association between dietary intake of vitamin D and the risk of developing RA.45 However, apart from supplements, the main source of vitamin D is de novo synthesis in the skin and estimated dietary intake may be a poor predictor of serum vitamin D concentrations. In a study of 79 patients with RA, no association was found between prior serum vitamin D concentrations and subsequent development of RA.46 However, it is notable that the geometric means for both cases and controls in this study were only half the lower reference level of 60 nmol/L, which has been set subsequently to reflect a level that suppresses secondary hyperparathyroidism due to vitamin D insufficiency.
Antioxidants and Risk of Rheumatoid Arthritis
Oxygen free radicals (e.g., nitric oxide, superoxide, hydroxyl radical) are implicated in the tissue damage observed in RA.47 Antioxidants including vitamin E (α-tocopherol), vitamin C (ascorbic acid), β-carotene, and selenium may have a protective role against tissue damage caused by these oxygen free radicals. This combined with evidence that markers of antioxidant nutritional status are lower in patients with established RA compared with normal controls48 led to the hypothesis that antioxidants may protect against the development of RA. Despite this biologic plausibility, the available data do not provide clear evidence for a protective effect of antioxidants as dietary supplements in relation to the development of RA.
Low serum concentrations of vitamin E, β-carotene, retinol, and selenium have been reported to be weakly associated with an increased risk for developing RA in some but not all studies.49–51 The strongest association between risk of RA and antioxidants was found for a combined antioxidant index rather than any one specific antioxidant.49
A higher dietary intake of β-cryptoxanthin (a carotenoid found in fruit and vegetables) and zinc may protect against the development of RA.52,53 Low vitamin C intake has also been associated with an increased risk of inflammatory polyarthritis with an adjusted OR of 3.3 (95% CI, 1.4 to 7.9) for the lowest tertile of vitamin C intake (<55.7 mg/day) compared with the highest intake tertile (>94.9 mg/day).54 However, another study found no association between intake of vitamin C, vitamin E, zinc, or selenium and the development of RA.31
The Women’s Health Study, a randomized, double-blind, placebo-controlled trial designed to evaluate the effects of low-dose aspirin and vitamin E in the primary prevention of cardiovascular disease and cancer, has been used to address the influence of vitamin E supplementation (600 IU on alternate days) on the development of RA. During the 10-year follow-up period, 106 cases of RA occurred with no significant association between vitamin E supplementation and incidence of RA. A statistically insignificant trend toward inverse association between vitamin E and development of seropositive RA was seen on subgroup analysis.55
Obesity and Rheumatoid Arthritis
Two studies have reported that obesity increases the risk of RA,56,57 whereas two other studies report no association.58–60 Increased plasma concentrations of the adipokines leptin, adiponectin, and visfatin have been observed in patients with RA compared with healthy controls.61 Furthermore, visfatin and leptin were associated with increased and reduced radiographic joint damage, respectively.61 These data combined with the proinflammatory state observed in obesity suggest that BMI may affect disease activity and outcomes in RA, whereas, perhaps paradoxically, increased BMI has been associated with less radiographic damage.58,62 An alternative explanation is that patients with more active inflammatory disease, who tend to develop more radiographic erosions, may have lower BMI and rheumatoid cachexia.
Gout
Key Points
The link between diet and gout has been recognized for centuries.
High intake of meat, seafood, and alcohol is associated with increased risk of gout.
Fructose increases serum urate.
Higher intake of low-fat dairy products has been associated with a reduced risk of gout.
Dietary Factors and Gout
Several large clinical studies have identified an association between high intake of meat and seafood (but not total protein intake) and both SU concentrations63 and gout.64 In comparison, the risk of gout is reduced with higher intake of low-fat dairy products and long-term coffee consumption.64,65 More recently, fructose, which is found in corn syrup, sugar-sweetened soft drinks, and fruit juices, has been associated with hyperuricemia and gout.66,67 Fructose increases SU through increased purine degradation.68 Furthermore, urate and fructose share a common transporter within the kidney (SLC2A9).69 Alcohol consumption other than moderate wine intake is associated with an increased risk of gout, with beer conferring a higher risk than liquor.70
Dietary vitamin C supplementation has been shown to reduce the risk of gout (relative risk of gout with no supplementation vs. 1000 to 1499 mg vitamin C/day, 0.66 [95% CI, 0.49 to 0.88]).71 Vitamin C (ascorbic acid) is an important vitamin that can only be obtained through dietary intake. Because ascorbic acid is water soluble, it is not stored within the body and thus must be regularly supplemented through the diet to maintain the ascorbic acid pool. Dietary sources of ascorbic acid include fresh fruits and vegetables, in particular citrus fruits and green leafy vegetables such as broccoli.
Fasting and Gout
Prolonged periods (2 weeks to 8 months) of fasting in obese subjects result in a significant increase in SU and in some cases the development of gout.72 In patients with previously documented gout, a 1-day fast led to an increase in SU of 0.5 to 2.1 mg/dL with a mean rise of 1.1 mg/dL, with refeeding associated with a return in SU to baseline levels after 24 hours.73 Explanations for the increase in SU during fasting include increased urate production, decreased excretion due to decreased glomerular filtration rate, altered renal tubular transport of uric acid, and competition with ketones for renal tubular excretion.74
Obesity and Gout
High BMI predisposes to gout.75 In a study comparing obese men (mean BMI 34 ± 4 kg/m2) with healthy controls (BMI, 21 ± 1 kg/m2), SU concentrations were raised to a similar degree in the obese subjects (≈8.0 ± 1.6 mg/dL vs. controls, 5.2 ± 0.81 mg/dL), irrespective of body fat distribution (predominantly visceral or predominantly subcutaneous). By contrast, 80% of subjects with hyperuricemia and accumulation of fat subcutaneously had low 24-hour urinary urate excretion compared with 10% of their counterparts with fat accumulation predominantly in a visceral distribution. These data suggest that the mechanism of hyperuricemia may vary depending on body fat distribution.76 In a separate study using computed tomography to determine transverse area of abdominal fat components, visceral fat was shown to correlate strongly with SU, whereas no relationship was seen between SU and BMI or subcutaneous fat area.77
Osteoarthritis
Key Points
Obesity is associated with knee osteoarthritis (OA).
Direct biomechanical effects of obesity contribute to OA.
Increased leptin provides another link between obesity and OA.
Associations have been established between obesity and onset and progression of knee OA. The evidence for an association between obesity and OA of the hip or hand is less well defined. In addition to direct biomechanical effects of obesity on the joint, recent data suggest a role for the adipocytokine leptin. Expression of leptin was increased in advanced OA cartilage compared with minimally damaged OA cartilage, with the leptin mRNA expression in advanced OA cartilage correlating with BMI. Furthermore, leptin was shown to reduce chondrocyte proliferation and increase IL-1β, MMP-9, and MMP-13 expression.78
Dietary antioxidants (vitamin E, vitamin C, and β-carotene) have been reported to reduce progression of knee OA but have no effect on onset of OA.79,80 The association between serum vitamin D concentrations and OA remains unclear, with one study reporting no association between vitamin D and risk of joint space narrowing or cartilage loss in knee OA81 and another study reporting a positive association between knee cartilage volume and serum vitamin D concentrations.82
Nutrition in the Management of Rheumatic Diseases
The role of nutrition in the management of gout is well accepted. However, its role in other rheumatic diseases such as RA is less routine. Despite a relative lack of interest or emphasis from physicians, many patients consider food may contribute to their arthritis and seek information about or try putative dietary remedies. A convincing, informed approach to nutritional advice is thus an important aspect of management, which can help patients avoid worthless interventions that are expensive, time consuming, in some cases harmful, and diversions from access to more effective measures (summarized in Figure 68-3). Furthermore, positive advice regarding dietary choices may empower the patient at a time when there is often a sense of loss of control. Well-informed, authoritative dietary advice can also protect patients from poorly grounded advice from relatives, friends, and nonauthoritative Internet sites about dietary measures.
< div class='tao-gold-member'>
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


