29 Neural Regulation of Pain and Inflammation
For over 2000 years (since Celsus and Galen), clinicians recognized that cardinal features of neurogenic responses, such as redness, warmth, swelling, and pain, are rapid sequelae of inflammation. Neurogenic vasodilatation reported in 1876 by Stricker and in 1901 by Bayliss1,2; the inflammatory axon reflex with erythema observed in the 1910s by Bruce and by Breslauer3,4; the flare response reported by Lewis around 1930 with erythema, hyperalgesia, and edema5; rediscovery of the antidromic vasodilatory flare response and dorsal root reflex by Chapman6; and Kelly’s and Jancsó’s more extended concepts of neurogenic inflammation in the 1960s7,8 all were expressions of the same principle: the influence of sensory afferent nerve fibers on acute inflammation and on cardinal clinical signs of inflammation. In the past two decades, our view has expanded to include the sympathetic and parasympathetic efferent nervous systems in inflammatory/immune control.
The concept of neuronal regulation of inflammation is supported by reports of patients with hemiplegia and chronic inflammatory diseases, in whom the paralytic side is protected from inflammation (Table 29-1). Cases have been reported in which hemiplegia manifested long after the outbreak of chronic inflammatory disease or long before, leading to protection independent of the time point (see Table 29-1).
Table 29-1 Role of Neuronal Innervation in the Development of Rheumatoid Arthritis and Other Inflammatory Diseases
| Situation | Modulation of Disease Symptoms | References |
|---|---|---|
| Poliomyelitis paralysis | RA only on the nonparalyzed side | 244 |
| Hemiplegia | RA only on the nonparalyzed side | 245–257 |
| Hemiplegia | RA vasculitis only on the nonparalyzed side | 258 |
| Hemiplegia | Gout only on the nonparalyzed side | 259 |
| Hemiplegia | Skin changes in PSS only on the nonparalyzed side | 260 |
| Hemiplegia | Psoriatic arthritis only on the nonparalyzed side | 261 |
| Sensory denervation | Denervated finger is spared from psoriatic arthritis | 262 |
| Brachial plexus lesion | Shoulder inflammation in a PMR patient only on intact side | 263 |
| Hemiplegia | DTH skin lesions more marked on the nonparalyzed side | 264 |
| Hemiplegia | Hemochromatosis arthritis only on the nonparalyzed side | 265 |
DTH, delayed-type hypersensitivity; PMR, polymyalgia rheumatica; PSS, progressive systemic sclerosis; RA, rheumatoid arthritis.
In addition, clinical observations demonstrate neuronal regulation of inflammation in that symptoms of many chronic inflammatory diseases have diurnal variation, with greater activity in the night and early morning hours. Because the rhythm of circadian changes in clinical signs depends on superordinate control of the hypothalamic nucleus suprachiasmaticus, a functional connection is revealed between the central nervous system (CNS), efferent pathways of the CNS (hormonal and neuronal), and the inflammatory/immune response.9
Neuronal regulation of inflammation is dependent on a robust innervation of lymphoid organs and the direct influence of neurotransmitters/neuropeptides on immune cells. Although sympathetic nerve fibers usually follow arteries (also branching into vessel-free regions), sensory nerve fibers have their own routes along vessels or independent of the vasculature. In addition, nerve fibers of the parasympathetic nervous system innervate many tissues in the head, neck, and trunk of the body, and upper and lower limbs are excluded. The greatest support for a neuroimmune contact comes from innervation of lymphoid organs, where nerve fibers are responsible for neuronal regulation of immune responses.10–12
The receptors for neurotransmitters are present on almost all immunocompetent cells. Some exceptions are known, such as the absence of β2-adrenergic receptors on T helper type 2 cells.13 The differential and time-dependent expression of receptors can shape the neuroimmune cross-talk. Sometimes receptor expression is increased or decreased in the context of an inflammatory response.14–16 The time-dependent involvement of different immune cells and receptor expression in the course of a given disease is probably important for neuronal regulation of inflammation.
In addition, intracellular signaling pathways of neurotransmitter receptors are dependent on environmental conditions; this has been demonstrated for G protein–coupled receptors.17,18 Another control process of these receptors involves regulators of G protein signaling (RGS), and tumor necrosis factor (TNF) can lead to increased desensitization of Gα protein–coupled receptors.19 For additional details on some receptors of neurotransmitters on immune cells, the reader is referred to the literature.13,20
In the study of neuronal regulation of inflammation, the bipolar role of neurotransmitters is important. For example, norepinephrine binds to α- and β-adrenergic receptors, which exert opposing effects on intracellular signaling cascades (α1: increase in diacylglycerol and protein kinase C; α2: decrease in cyclic adenosine monophosphate [cAMP]; β: increase in cAMP). Although norepinephrine binds to α-adrenergic receptors at between 10−9 and 10−5 mol/L, it binds to β-adrenergic receptors only at concentrations equal to or higher than 10−7 mol/L (typical serum level: 10−9 mol/L; typical tissue concentration: 10−7 mol/L). Because α-adrenergic effects (proinflammatory) are markedly different from β-adrenergic effects (anti-inflammatory), the concentration of norepinephrine in the environment of an immune cell is very important for noradrenergic effects. The situation is very similar for adenosine via A1 adenosine receptors (e.g., α-adrenergic) and A2a/b adenosine receptors (e.g., β-adrenergic). It is important to note that this behavior is typical for many neurotransmitters/neuropeptides because more than one receptor can be a binding partner, and different receptors have opposing intracellular signaling pathways.21 In conclusion, neuronal regulation of inflammation is an important aspect of the inflammatory process.
During evolution, mechanisms were positively selected that serve to overcome acute transient inflammatory episodes but not chronic lifelong inflammation, because of the negative selection pressure.22,23 Transient inflammatory episodes, for example, include infections, wound healing responses, foreign body reactions, immune reactions during pregnancy, and others.22,23 Mechanisms of these short-lasting episodes are also used in chronic inflammatory diseases. From this point of view, it is meaningful to start with an acute transient inflammatory episode such as a wound response after injection of foreign material into the skin.
Acute Inflammation (The First 12 Hours)
Recognition of Foreign or Pathogenic Material: Immune and Pain Pathways
Systemic recognition of foreign material occurs in highly specialized nerve endings of sensory afferent, nociceptive nerve fibers (the nociceptor). Nerve endings of sensory afferent nerve fibers possess an impressive array of receptors that are responsible for instant activation of the nerve fiber (Figure 29-1).24,25 Upon introduction of foreign material, infectious agents can pose a threat, which can elicit a neuronal response via pattern recognition receptors on polymodal nociceptors (e.g., the Toll-like receptors) (see Figure 29-1). In addition, factors such as bradykinin, prostaglandins, and cytokines from activated mast cells and other cells can stimulate their respective receptors on sensory nerve terminals (see Figure 29-1). Under consideration of these mechanisms, peripheral recognition of foreign material by nociceptors is part of the innate immune response. Moreover, mechanical irritation, noxious cold/heat, and low pH concentration stimulate the sensory afferent nerve fiber (see Figure 29-1). Altogether, this leads to an orthodromic action potential that stimulates the dorsal root ganglion (DRG) and releases elements such as substance P into the wounded peripheral tissue (efferent function of sensory afferents). The spreading reaction is attributed to the axon reflex and the dorsal root reflex, which lead to antidromic activation of neighboring sensory afferents, resulting in local expansion of the immediate flare response.24,26,27
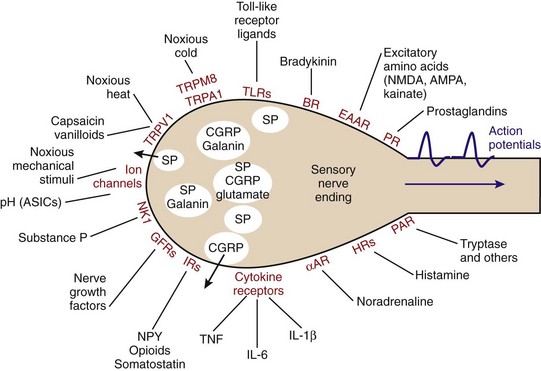
Substance P is one of the strongest chemotactic and vasodilatory factors; it leads to instant plasma extravasation and accumulation of neutrophils, monocytes, and other cells.28–30 Substance P and other neuropeptides increase vascular leakage and interstitial fluid volume in connective tissue capsules, tendons, and muscles, leading to stiffness. In addition, substance P immediately stimulates activities of mast cells, monocytes, macrophages, dendritic cells, and neutrophils to reflexively increase local proinflammatory responses. Parallel to substance P, calcitonin gene–related peptide (CGRP) with strong vasodilatory and chemotactic activities is released. The third sensory neurotransmitter is the excitatory amino acid glutamate, whose proinflammatory effects have been described.31 The fourth neurotransmitter of sensory afferent nerve fibers is galanin, which possibly has dual proinflammatory and anti-inflammatory roles, depending on receptor subtypes (but data are limited with respect to effects on immune cells).32–34 All these neurotransmitters/neuropeptides are locally secreted into the vicinity of the peripheral nerve terminal.
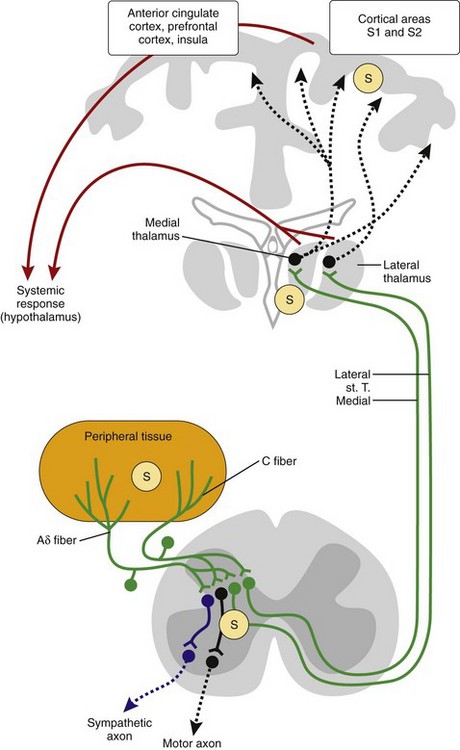
In addition to local effects of these neurotransmitters/neuropeptides, pain signals are transmitted to the brain and elicit a systemic response (Figure 29-2). The pathways ascend through sensory nerve fibers (Aδ or C fibers), the neurons in the DRG, the neurons in the spinal medulla, and the contralateral spinothalamic tract to reach the medial and lateral thalamus, cortical areas S1 and S2, the hippocampus, and other brain regions responsible for affective components of pain (anterior cingulate cortex, insula, and prefrontal cortex)35 (see Figure 29-2). All parts of the pain pathway can be sensitized under the influence of inflammatory stimuli. Sensitization means stabilization and amplification of nociceptive stimuli.
Peripheral Sensitization
Sensitization appears already during the earliest phase of inflammation, as demonstrated in the kaolin/carrageenan or similar instant chemical models. Nevertheless, sensitization is a dynamic process that changes over time, as demonstrated by inflammation-induced induction of transient receptor potential vanilloid-1 (TRPV1) receptors on DRG neurons, gradual infiltration of macrophages into the DRG, or bilateral long-term upregulation of bradykinin receptor B2 in the DRG and dorsal horn.36–40 Thus, sensitization plays a role throughout all inflammatory phases, but the underlying mechanisms might change over time.
In normal tissue, nociceptors have high thresholds. However, during inflammation, these thresholds are lowered and nociceptors are sensitized.25,41 Lowering of the nociceptor threshold is a consequence of converging stimulatory inputs into the nerve terminal via different receptor pathways (see Figure 29-1). These high-threshold units, defined as nociceptors by their high mechanical threshold, become sensitized and start to respond to light pressure and movements in the working range of the joint (Figure 29-3A). Most of these units are thin myelinated fibers (Aδ fibers) or unmyelinated fibers (C fibers). Furthermore, mechanoinsensitive and thermoinsensitive “silent” nociceptors are sensitized in inflamed tissue, and they start to respond to mechanical and thermal stimuli during inflammation.25,41 This class of receptors is characterized by long-standing responses to algogenic factors, and they are important in neurogenic inflammation.25,42 These mechanisms are summarized under the heading of peripheral sensitization (the “S” in Figure 29-2).
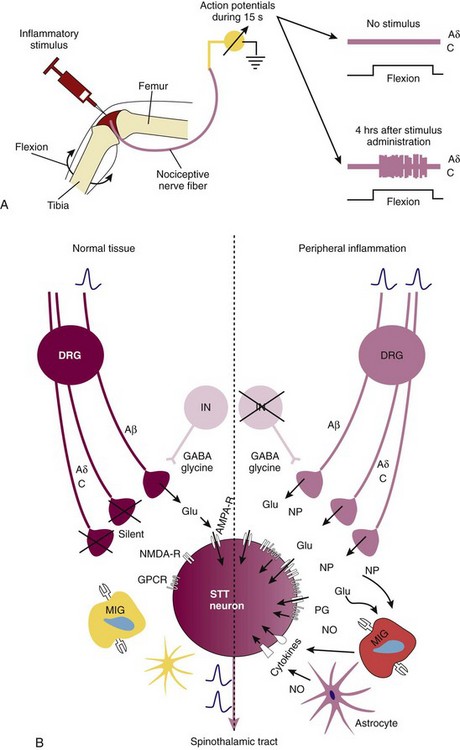
Figure 29-3 Mechanisms of peripheral and central sensitization. A, Peripheral sensitization. Injection of an inflammatory stimulus leads to an increase in the number of action potentials as recorded from afferent fibers supplying the joint.13,14 Peripheral sensitization is mediated by a plethora of heterogeneous receptors on afferent fibers (see Figure 29-1). B, Spinal central sensitization. Left, In the normal situation, only Aβ fibers are activated (upon mechanical stimuli); these are low-threshold nonnociceptor fibers that release glutamate (Glu). On the postsynaptic neuron, only α-amino-3-hydroxyl-5-methyl-4-isoxazole-4-propionic acid (AMPA) receptors are activated and opened.13,14 Right, In the inflammatory situation, previously high-threshold Aδ and C fibers are activated by pressure, leading to release of glutamate and neuropeptides (NPs) such as substance P and calcitonin gene-related peptide (CGRP). This leads to activation of the postsynaptic membrane via AMPA (AMPA-R) and N-methyl-d-aspartate (NMDA) receptors (NMDA-R), neuropeptide receptors, prostaglandin receptors, and cytokine receptors (particularly, IL-1β, IL-6, TNF). These changes lead to long-standing hypersensitivity. DRG, dorsal root ganglion; GABA, γ-aminobutyric acid; GPCR, G protein–coupled receptor; IN, interneuron; MIG, microglia; NMDA, N-methyl-d-aspartate; NO, nitric oxide; NP, neuropeptide; PG, prostaglandin; STT, spinothalamic tract. The star-formed cell is an astrocyte.
It is important to note that injection of proinflammatory cytokines such as interleukin (IL)-6 and TNF into the joint leads to a huge increase in the number of action potentials recorded from afferent fibers supplying the joint. Both IL-6 and TNF have the potential to sensitize afferent nerve fibers in the joint to mechanical stimulation contributing to mechanical hypersensitivity.43,44 These effects can be blocked by anticytokine therapy with biologic agents,43,44 and it is expected that this also inhibits release of proinflammatory substance P and other neuropeptides.
Central Sensitization
In the DRG and spinal cord, peripheral inflammation makes neurons hyperexcitable and more susceptible to input from sensory nerve fibers (the “S” in Figure 29-2). This amplifies the response through additional activation of adjacent and even remote spinal neurons far away from the inflamed region, leading to expansion of the receptive field.25,41 The peripheral inflammatory response increases expression of substance P, CGRP, and bradykinin with their respective receptors in the DRG and dorsal horn.45,46 In the dorsal horn, substance P potentiates the release of factors such as glutamate and aspartate.47 The ipsilateral response can lead to contralateral co-activation of the DRG and sensory afferents,36 which might contribute to symmetric manifestations of inflammation.48,49 Bilateral upregulation of, for example, neurokinin 1 and bradykinin 2 receptors has been demonstrated, whereby this phenomenon was strictly segmental and not general.36 Sometimes spinal sensitization persists beyond the peripheral nociceptive or inflammatory process, and the character of pain changes from an inflammatory to a neuropathic form.50 In experimental arthritis, such a shift from inflammatory to neuropathic features of sensitization has been demonstrated by increased expression of a typical marker of neuropathic pain, ATF3, and by the favorable effects of gabapentin treatment in a postinflammatory phase of hypersensitivity.50
Spinal sensitization is often a consequence of increased release of excitatory amino acids (glutamate, aspartate, and glycine), substance P, CGRP, neurokinin A, and galanin from nociceptor neurons and upregulation of the respective receptors in the spinal medulla. Enhanced release can be induced by peripheral inflammation.24,25,41,51 Non–N-methyl-d-aspartate (NMDA) receptors but also NMDA glutamate receptors are relevant in joint inflammation (Figure 29-3B).52,53 Sensitization can be mimicked experimentally by intrathecal administration of substance P or NMDA via an increase in prostaglandins or cyclooxygenase-2.54 In addition to the activating pathway, there exist inhibitory pathways via, for example, γ-aminobutyric acid (GABA) or glycine.55 Second, spinal sensitization is dependent on microglial cells and astrocytes, which can aggravate pathologic pain states in which cytokines and chemotactic factors play an important priming and perpetuating role (see Figure 29-3B).56,57 Cytokines such as IL-1β, IL-6, and TNF play a dominant role in cytokine-induced hypersensitivity,56,57 and these cytokines are induced in the spinal cord during experimental arthritis.58
Several proinflammatory intracellular signaling pathways have been implicated in priming of microglia and pain-processing neurons. It is not easy to distinguish whether signaling cascades in neurons, microglia, or other cells are important, because experimental studies using microdialysis or intrathecal administration of pathway inhibitors do not target a specific cell type. Nevertheless, these studies clearly demonstrate the importance of factors such as nuclear factor κB (NFκB),59 protein kinase A,60 protein kinase C,61,62 c-Jun N-terminal kinase,63 JAK/STAT3 signaling pathway,64 p38 mitogen-activated protein kinase (MAPK),65–67 Src-family kinase,68 arachidonic acid pathways,69 and others. These pathways typically lead to intraneuronal calcium and sodium accumulation, which is an excitatory signal.24,41
For example, the p38 pathway was demonstrated to be an important proinflammatory signaling cascade in spinal neurons and microglial cells in experimental arthritis.65 Phosphorylated p38 is increased in microglial and neuronal cells during the course of experimental arthritis. Intrathecal administration of a specific p38 inhibitor led to decreased synovial inflammation but also to suppressed articular cytokine and protease expression and joint destruction as measured by radiographic and histology scores.65 This effect was dependent on the presence of TNF in the spinal cord. TNF can be a signaling element upstream of p38 by activating p38-phosphorylating kinases, or downstream of phosphorylated p38 that induces TNF secretion. It was demonstrated that intrathecal, but not subcutaneous, TNF neutralization with etanercept inhibited p38 phosphorylation and peripheral inflammation.65 The positive effect of intrathecal spinal TNF neutralization was confirmed in another model of experimental arthritis.70 In this model, peripheral joint inflammation was decreased, and pain-related behavior was drastically reduced.70
It is interesting that all mentioned pathways belong to proinflammatory cascades initially described in peripheral immune cells. In contrast, anti-inflammatory pathways such as adenosine A1, β-adrenergic, and δ/µ-opioidergic receptor pathways are inhibitory in microglial activation paradigms.71–75 For instance, spinal administration of an A1 adenosine receptor agonist markedly reduced inflammation, as well as bone and cartilage destruction, in an experimental arthritis model.71,72 Administration of the A1 adenosine receptor agonist also decreased nuclear c-Fos expression in the superficial and deep dorsal horns of the spinal medulla. In addition, the A1 adenosine receptor agonist decreased the density of astrocytes in these areas.72 This indicates that, along with neurons and microglial cells, astrocytes are involved in sensitization.
Finally, understanding why peripheral and central sensitization was conserved during evolution is important. Sensitization of pain has a protective role because it warns about potential danger, enables us to remove noxious stimuli, and stimulates wound management. Furthermore, avoidance of painful situations in the future would be desirable. This is nicely indicated by the fact that peripheral inflammatory stimulation of sensory neurons can induce central IL-1β release in the hippocampus,76 a cytokine that is instrumental in hippocampal learning phenomena.77 Sensitization is an amplification factor that should last as long as painful or noxious stimuli are present (or even a little longer to stimulate wound management). Thus, sensitization is a supportive factor of innate immunity. It has been positively selected as an evolutionarily conserved learning phenomenon that will not be stopped until inflammation is terminated (i.e., the stimulus is removed).
Neuroendocrine Systemic Response
Parallel to the local inflammatory reaction, hormonal and neuronal systemic responses are engaged. The hormonal response system is mainly the hypothalamic-pituitary-adrenal (HPA) axis, which is stimulated through activation of sensory pathways or through circulating cytokines.78 Activation of the HPA axis can happen on adrenal, pituitary, and hypothalamic levels.78 Neuronal efferent response systems include the sympathetic nervous system and the parasympathetic nervous system.
The systemic response of the HPA axis and the sympathetic nervous system is an “energy appeal reaction” that serves allocation of energy-rich fuels from stores to the highly catabolic immune system (Figure 29-4).79,80 This re-allocation of energy-rich fuels is important throughout the inflammatory response. The energy demand of the immune system can contribute to a systemic response whose long-standing use is detrimental to homeostasis (see Figure 29-4).79,80
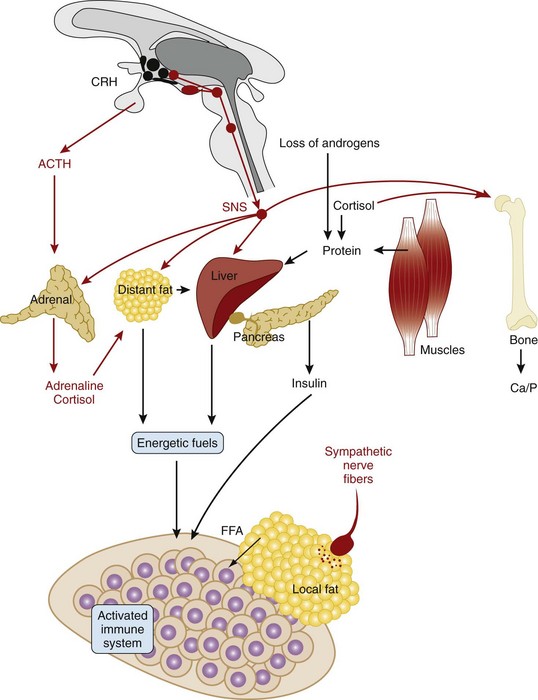
Figure 29-4 Systemic changes in the nervous system during systemic inflammation. Peripheral inflammation activates the hypothalamus, leading to increased activity of the sympathetic nervous system (SNS) and the hypothalamic-pituitary-adrenal (HPA) axis. Upregulation of the SNS leads to different reactions that re-allocate energy-rich fuels to the activated immune system (fat tissue: β-adrenergically mediated lipolysis; liver: β-adrenergically and cortisol-mediated gluconeogenesis; muscle: cortisol-mediated and androgen loss–induced muscle breakdown leads to protein provision for gluconeogenesis). This is accompanied by a breakdown of bone (via cortisol, via increased sympathetic activity, via inflammatory pathways). All these consecutive reactions of inflammation are important to provide energy-rich fuels to the immune system. Loss of sympathetic nerve fibers in the center of inflamed tissue is discussed in Figure 29-5. The parasympathetic nervous system is downregulated and is not shown. ACTH, adrenocorticotropic hormone; Ca, calcium; CRH, corticotropin-releasing hormone; FFA, free fatty acid; P, phosphorus.
In the course of inflammation, the sympathetic nervous system is activated via direct spinal interneurons that link sensory inputs to sympathetic output (see Figure 29-2, blue lines). This has the advantage that the input defines the location of the output, which leads to confinement of the response to the affected area. In addition to the central coupling of sensory and sympathetic pathways, sympathetic nerve fibers communicate with sensory nerve terminals by way of α2-adrenergic and prostaglandin cross-signaling at the level of the peripheral nerve terminal.81,82 This inflammation-induced cross-signaling leads to higher activity of sensory afferents. Systemically, the sympathetic nervous system, similar to the HPA axis, is activated through stimulation of sensory pathways or through circulating cytokines (e.g., IL-6). Activation of the sympathetic nervous system is the main factor in re-allocating energy to the immune system (see Figure 29-4).
Although systemically relevant inflammation is coupled to increased sympathetic nervous tone and increased activity of the HPA axis (albeit inadequately low in relation to inflammation) (see Figure 29-4), the activity of the parasympathetic nervous system and the hypothalamic-pituitary-gonadal (HPG) axis is inhibited.83,84 This leads to the well-known dissociation of sympathetic versus parasympathetic and HPA axis versus HPG axis activity (androgens are low), respectively. This serves the re-allocation of energy-rich fuels to the immune system (see Figure 29-4).79,80
Activation of the HPA axis and the sympathetic nervous system at the onset of inflammation prepares the immune system for most naturally occurring immune challenges.85 Activation of the HPA axis mobilizes immune cells, leading to redistribution of neutrophils, monocytes, and natural killer (NK) cells.85 The sympathetic nervous system can support the very acute inflammatory process in phase 1 because of six main mechanisms: (1) mobilization of immune cells from systemic stores (similar to the HPA axis),85 (2) support of plasma extravasation,86 (3) remodeling of tissue by inducing matrix metalloproteinases,87,88 (4) stimulation of nociceptors via α2-adrenergic and prostaglandin cross-signaling,81,82 (5) chemoattractant activity of sympathetic neurotransmitters,89 and (6) liberation of free fatty acids and glucose necessary for the activated immune system (see Figure 29-4). In summary, during the first hours of inflammation, the HPA axis and the sympathetic nervous system are mainly proinflammatory.
Vagal afferents from the intestine and liver play an important role in modulating a systemic milieu that increases or decreases the magnitude of very acute inflammatory hyperalgesia, which depends on the agent, the stimulus strength, and epinephrine secretion from the adrenal medulla.90,91 The vagal tonus determines the overall reflex modulation of very acute inflammatory processes, and vagal afferents are important in perception of inflammatory conditions in the abdomen.92,93 Reports in the last decade have demonstrated that lipopolysaccharide-induced inflammation can be inhibited by electrical vagus stimulation of the distal end of the dissected vagus nerve.94 These very acute vagal effects were dependent on the sympathetic innervation of the spleen.95 In addition, carrageenan-induced leukocyte recruitment into a pre-formed subcutaneous air pouch was inhibited by vagus nerve stimulation of the intact vagus nerve.96 This was done without dissection of the vagus nerve so that afferent and efferent vagus nerve effects cannot be separated.96
Administration of an intrathecal p38 inhibitor (mentioned earlier), which has favorable effects in experimental arthritis, largely increases vagal activity.97 Because spinal application of p38 inhibitors blocks aspects of central sensitization,65 one would expect blockade of segmental sympathetic outflow, as demonstrated in Figure 29-2 (blue lines). A decrease in central pain signaling, and thus decreased hypothalamic activation of the HPA axis and sympathetic nervous system, and diminution of segmental sympathetic outflow most probably increase parasympathetic reflex activity. Particularly in very early inflammation, this should be a favorable anti-inflammatory feature. These acute vagus experiments were complemented by experiments in long-standing chronic inflammation models (see later).
Intermediate Inflammation (Between 12 Hours and Several Days/Few Weeks)
Local Cell Accumulation in Inflamed Tissue
Local cells accumulate in inflamed tissue as a consequence of cell mobilization and chemotaxis. The major neurotransmitters/neuropeptides of sensory afferents (substance P) and of sympathetic efferents (norepinephrine) are potent chemotactic factors for innate immune cells, such as neutrophils, monocytes, and eosinophils. The direct chemotactic effect of substance P has been demonstrated by injecting substance P into the skin; this leads to upregulation of the endothelial adhesion molecule E-selectin (CD62E) and, for example, attraction of eosinophils to the injection site.98 Similarly for the sympathetic nervous system, the lack of catecholamine production in animals with a deletion of the dopamine-β-hydroxylase gene leads to a strong reduction of leukocyte accumulation in the adventitia and periadventitia of vessels.99
Substance P and norepinephrine also have strong chemotactic effects in vitro.89,100 The sympathetic co-transmitter neuropeptide Y and the sensory co-transmitter CGRP also have chemotactic effects.89,101 The effects of substance P and norepinephrine can be amplified by increasing secretion of potent chemotactic factors such as IL-8.102,103 In addition, norepinephrine and substance P can upregulate matrix metalloproteinases to soften the tissue.87,88,104
Immediate Change in Neuronal Innervation
Upon entry of monocytes and neutrophils into the tissue, these cells become activated and can engulf pathogens or foreign material. Activation of these cells is mediated by pattern recognition receptors, as well as by other proinflammatory mediators and inflammasome-derived products, leading to activation of neutrophils and differentiation of entering monocytes into macrophages or dendritic cells.105
In striking contrast, norepinephrine and its potent co-transmitter adenosine (made from sympathetically released adenosine triphosphate [ATP]) inhibit many proinflammatory effects of activated innate immune cells such as monocytes, macrophages, NK cells, and neutrophils via β-adrenergic and A2 adenosine receptor signaling.106 In this context, it is important to mention that ectonucleotidases (CD39 and CD73), which convert purine precursor neurotransmitters to adenosine, are increased in inflammation.107–109 A classic example of A2-adenosine–mediated or β-adrenergically induced inhibition of cells is the strong negative effect on neutrophil or monocyte/macrophage phagocytosis and on the function of dendritic cells. The important role of adenosine as an inducer of regulatory T lymphocytes has been carefully documented.110 Because innervation is balanced in tissue with similar densities of sensory and sympathetic nerve fibers, this dichotomy of substance P and norepinephrine/adenosine is counterproductive for innate immunity.
Activated macrophages and stimulated tissue fibroblasts start to produce nerve growth factor (NGF), which supports the outgrowth of sensory and sympathetic nerve fibers equally well. In other words, NGF is not specific for sensory or sympathetic nerve fibers. Indeed, inflammatory tissue releases large amounts of NGF, as, for example, is substantiated in rheumatoid arthritis (RA) or experimental arthritis.111,112
Activated macrophages and fibroblasts also produce nerve repellent factors such as semaphorin 3C and semaphorin 3F.113,114 These two factors specifically repel sympathetic nerve fibers and have no effect on sensory nerve fibers, which instead are repelled by semaphorin 3A.113,114 In addition, sensory nerve fibers sprout under the influence of NGF into inflamed tissue, leading to a preponderance of substance P over sympathetic neurotransmitters.115 Such a sensory hyperinnervation is also observed in skin wounds when sympathetic nerve fibers are absent.116 Loss of sympathetic nerve fibers is a rapid process that is observed soon after initiation of experimental inflammation.117–119 It can also be observed in vitro with the use of repellent factors in neurite outgrowth assays (within a few hours).114
Repulsion of sympathetic nerve fibers and sprouting of sensory nerve fibers are important ways to initiate a proinflammatory environment in the later phase of inflammation. As shown in Figure 29-5, the appearance of two noradrenergic zones (β: normal/healthy; α: inflamed tissue) is a consequence of this process. It is important to mention that loss of sympathetic nerve fibers is observed not only in inflamed tissue but also in the spleen117,118,120 and in the lymph nodes. In the former, loss of sympathetic nerve fibers is evident in the white pulp (T cell proliferation area); similarly, these fibers are not observed in B cell follicles.117,121 In the same animals, sympathetic nerve fibers sprout into the hilus area of the spleen and do not reach the distal white pulp (T cell) or follicles (B cell). Thus, a proinflammatory milieu is established in secondary lymphoid organs, as in peripherally inflamed tissue.
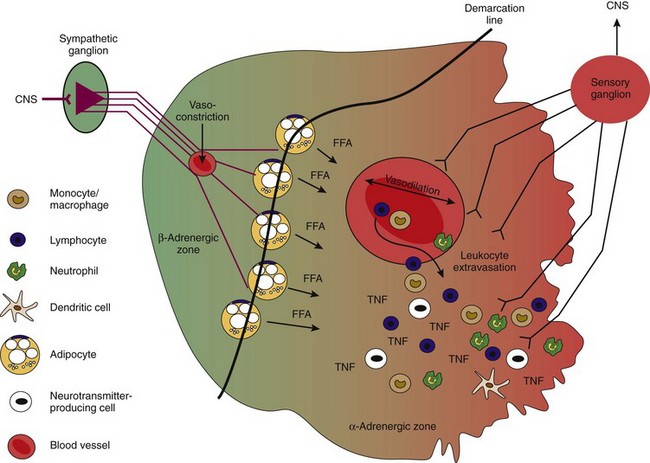
Role of Catecholamines in Antigen Transport to Secondary Lymphoid Organs and Immune Response
After antigen capture, a further important aspect of inflammation is the transport of processed antigenic material to secondary lymphoid organs. Transport to lymphoid organs is mediated by lymphatic vessels, whose pumping efficiency is decreased by β-adrenergic pathways and is stimulated by α-adrenergic signaling.122,123 In addition, migration of antigen-loaded dendritic cells is stimulated via α1-adrenergic mechanisms.124 It is important to note that immature dendritic cells migrate upon α1-adrenergic influence, but CD40-stimulated mature dendritic cells do not (those that arrived in secondary lymphoid organs and encountered T cell contact via CD40–CD40 ligand).124 Thus, rapid establishment of an α-adrenergic zone in peripheral tissue is probably important in inducing migration of dendritic cells. In addition, substance P supports dendritic cell maturation and activity.125,126
Catecholamine and its co-transmitter adenosine influence the direction of the immune response, whether T helper type 1 or type 2. Detailed in vitro experiments show that norepinephrine via β-adrenergic pathways inhibits T helper type 1 cell priming by inhibiting IL-12 and stimulating IL-10 of dendritic cells.20,127 The effects of catecholamines on the T helper type 17 immune response are not known. Tolerogenic effects of adenosine have been described.110 In addition to reactions on T cells, norepinephrine inhibits antigen presentation by epidermal Langerhans cells; this event is β-adrenergically mediated.128 Already in the late 1980s, it was demonstrated that surface expression of the antigen-presenting molecule human leukocyte antigen (HLA) class II was inhibited by β-adrenergic signaling.129,130
In summary, the sympathetic nervous system has many inhibitory roles via β2-adrenergic and A2-adenosine receptors when T helper type 1 cell priming participates (e.g., in arthritis). The opposite occurs in a situation with T helper type 2 conditions because norepinephrine stimulates IL-4 and IL-10; this occurs along with many stimulating effects on B cells and antibody production (e.g., in systemic lupus erythematosus).20 Because norepinephrine and adenosine have additional strong inhibitory effects on secretion of TNF, interferon (IFN)-γ, and IL-2 via β-adrenergic and A2 adenosine pathways, the general inhibitory aspect of these neurotransmitters at high concentrations, along with vasoactive intestinal peptide (VIP) and CGRP, is well established.131,132
At low concentrations of norepinephrine, when α1/2-adrenergic signaling is dominant, even stimulating effects on TNF occur.133,134 Thus, β-adrenergic influence in peripheral tissue and in secondary lymphoid organs should be reduced during proinflammatory T helper type 1 cell priming. Similar sympathetic nerve fiber loss and establishment of distinct α- and β-adrenergic zones belong to an adaptive process in secondary lymphoid organs to support or inhibit immune responses toward distinct antigens.
Clonal Expansion of Aggressive and Regulatory T and B Cells
The antiproliferative effects on T cells of norepinephrine via β-adrenergic receptors have been documented by many investigators.20,135 The proliferative response of CD8+ T cells is inhibited to a greater extent than that of CD4+ T cells, presumably because CD8+ T cells have a greater number of β-adrenergic receptors, and this effect is mediated via inhibition of IL-2 secretion.135 A proliferative effect of norepinephrine via β-adrenergic receptors is known for B cells.20,136,137 Similarly, the proliferative effect of substance P on T and B cells is common knowledge. The supportive effect of norepinephrine on antibody production has been demonstrated many times.20,136,138 These dichotomous effects of norepinephrine shape the immune response induced by T helper type 1 or type 2 priming antigens or autoantigens.
Resolution of Inflammation and Tissue Repair
Upon clearance of a pathogen, resolution of inflammation or reconstitution of normal tissue is the final step. Inflammation often leads to a preponderance of sensory nerve fibers (sensory hyperinnervation) over sympathetic nerve fibers, which are reduced in inflamed areas.115 In acute wounds, both nerve fibers disappear, but reappearance of sensory nerve fibers seems to start earlier than reinnervation with sympathetic nerves (Table 29-2).31,116,117,139–150 In general, reinstallation of sympathetic nerve fibers is a very long process, as substantiated in transplanted organs (>4 weeks), after tibial nerve crush (8 to 12 weeks), after chemical sympathectomy in the spleen (3 to 8 weeks), and after monophasic arthritis (4 to 8 weeks).151–154
Table 29-2 Behavior of Nerve Fibers and Their Neurotransmitters/Neuropeptides in Wound Reactions*
| Nerve Fiber Type | Change during Wound Reactions | References |
|---|---|---|
| Sensory nerve fibers | Sensory nerve fibers are lost after 2 days but reappear after approximately 7 to 14 days | 266–268 |
| Substance P and calcitonin gene-related peptide promote wound healing | 269–273 | |
| Sympathetic nerve fibers | Fast loss of sympathetic nerve fibers and reappearance after approximately 14 days | 274 |
| Catecholamines block wound repair via β-adrenoceptors | 275, 276 | |
| Norepinephrine inhibits wound macrophages and neutrophils | 277, 278 | |
| Catecholamines support later re-epithelialization | 279–282 | |
| Adenosine supports the wound healing response via A2 receptors (mediated through increase in fibrosis) | 283, 284 |
* Experiments with 6-hydroxydopamine, the sympathetic nerve fiber toxic substance, are not included because this substance affects not only sympathetic nerve fibers.
In a typical wound reaction, substance P promotes wound healing responses, and catecholamines have negative and positive effects on wound healing, such as inhibition of wound macrophages/neutrophils but support of later re-epithelialization (see Table 29-2). Moreover, stressful events that release sympathetic neurotransmitters and glucocorticoids lead to wound healing problems.155,156 From this point of view, a preponderance of substance P–positive nerve fibers over sympathetic nerve fibers would be favorable. Sensory hyperinnervation is probably supportive.
Chronic Inflammatory Disease
Neuronal Influences on Chronic Inflammatory Disease in Animal Models
Chronic inflammatory disease occurs when inflammation fails to resolve and tissue repair is inadequate. The neuronal elements can contribute to this process. Most studies investigated the role of the sympathetic nervous system in the adjuvant arthritis model in Lewis rats.157
The proinflammatory role of substance P and sensory nerve fibers in this model was demonstrated early in the 1980s.139 Additionally, substance P is proinflammatory for human synoviocytes and monocytes.158 Nociceptive fibers in the draining dorsal lymph nodes must play a critical role during this induction phase because local capsaicin treatment of these lymph nodes markedly decreases disease severity.159 Although the proinflammatory effects of substance P and other tachykinins are widely known, substance P–antagonistic therapies were not effective; this is probably a result of the redundancy in the tachykinin system.
In experiments conducted to study the role of the sympathetic nervous system in adjuvant-induced arthritis, most studies focused on a period of 14 to 40 days. From these studies, it is evident that overall peripheral sympathectomy or blockade of adrenoceptors (particularly via β-adrenoceptors) before or at the time of injection of Freund’s adjuvant diminishes the severity of joint inflammation during the entire observation period of 40 days.160,161 Similarly, the sympathetic nervous system plays an aggravating role in the collagen-induced arthritis (CIA) model, which is an autoantigen-driven chronic inflammatory disease. This might result from increased CD4+CD25+FoxP3–T cells, as was recently demonstrated.162 When the sympathetic nervous system was destroyed before immunization and up to day 18 after immunization, sympathectomy markedly reduced the severity of arthritis.162,163 However, when the sympathetic nervous system was destroyed after outbreak of the disease, sympathectomy strongly aggravated arthritis.163
The surprising dual role of the sympathetic nervous system might be explained as follows. In the very early induction phase after injection of adjuvant/antigen, mobilization, migration, and chemotaxis of proinflammatory cells such as neutrophils, NK cells, and monocytes directed to the site of adjuvant/antigen injection play a dominant role. In addition, targeted destruction of sympathetic nerves in secondary lymphoid organs supports antigen presentation and the switch to an aggressive effector immune response.161 Under conditions with sympathectomy in secondary lymphoid organs, arthritis becomes more severe owing to improved antigen presentation, stronger T helper lymphocyte type 1 immune reactions (aggressive phenotype for tissue-specific autoantigens), and probably downregulation of several regulatory elements such as IL-4 and IL-10 (see phase 2).161 Because the effect of prior sympathectomy is long lasting, these initial events are important for later inflammatory disease, which has also been demonstrated in atopic dermatitis and experimental colitis.164,165 However, in the chronic phase of the disease, the influence of the sympathetic nervous system largely changes.
One of the main changes is loss of sympathetic nerve fibers in inflamed tissue and in secondary lymphoid organs, as was already discussed (phase 2, Figure 29-5). Nerve fiber loss, starting with onset of disease,117,118,166 turns the essentially anti-inflammatory influence of sympathetic neurotransmitters at high concentrations into a proinflammatory influence at low concentrations (see Figure 29-5). In addition, recently described tyrosine hydroxylase–positive cells with anti-inflammatory capacities appear in lymphoid organs and arthritic tissue.167–169 In chronic inflammatory disease, the number of these cells in secondary lymphoid organs increases over time, and they appear shortly after disease outbreak in the inflamed joint.169 Because these cells can be eliminated by 6-hydroxydopamine treatment (the sympathectomy technique), the anti-inflammatory influence of these cells is soon lost after experimental chemical sympathectomy.163 Loss of tyrosine hydroxylase–positive cells probably leads to an overall proinflammatory situation, because these cells might have tolerogenic activities.170 The quite different effects of this sympathectomy tool are now explained by early destruction of sympathetic nerve fibers, which are proinflammatory (phase 1), and later destruction of anti-inflammatory catecholamine-producing cells in phase 3, which are anti-inflammatory.
Finally, the influence of the parasympathetic nervous system has attracted increasing interest. The alpha7 subunit of the nicotinergic acetylcholine receptor is especially relevant in the regulation of inflammatory responses171; this led to the concept of the cholinergic anti-inflammatory reflex. Additional experiments with agonists of the alpha7 subunit of the nicotinergic acetylcholine receptor demonstrated the anti-inflammatory importance of this cellular pathway in animal experiments and human cells.172–178 It is important to note that this particular nicotinergic receptor is highly expressed on macrophages and fibroblasts of patients with RA.175,177
At the moment, it is unclear how the vagus nerve influences synovial inflammatory disease. Four different possibilities exist regarding how favorable cholinergic effects on joint inflammation can be explained: (1) The cholinergic influence supports sympathetic inhibition of splenic proinflammatory immune responses, (2) the cholinergic influence directly affects cells in draining lymph nodes of the trunk, (3) the cholinergic influence affects cells in the gut (which play an important role as substantiated in the HLA-B27 rat model of arthritis), and (4) nonneuronal acetylcholine release appears within inflamed synovial tissue.118,178,179
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


