Fig. 1
Normal aged cartilage is biomechanically different from normal young cartilage. Over time, the surface (STZ: superficial tangential zone) becomes rougher and there are fewer chondrocytes. Degenerative aged and/or arthritic cartilage has deep surface clefts and fibrillation with chondrocyte clustering. In this diagram, the degree of gray shading indicates biomechanical integrity and loss of glycosaminoglycan (GAG). The table shows the location of changes in the STZ, middle zone (MZ), and deep zone (DZ). (•) indicates the full presence of changes, (ο) the complete absence, with the arrow indicating a change between cartilage states (Reprinted from Osteoarthr Cartilage, Vol. 17, Temple-Wong MM, Bae WC, Chen MQ, Bugbee WD, Amiel D, Coutts RD, Lotz M, Sah RL. Biomechanical, structural, and biochemical indices of degenerative and osteoarthritic deterioration of adult human articular cartilage of the femoral condyle, 1469–1476, Copyright (2009), with permission from Elsevier)
Although osteoarthritis is not an inflammatory process in the same sense as the rheumatologic diseases that cause joint destruction, inflammation is clearly part of what causes radiographic osteoarthritis to become painful. When osteoarthritis becomes symptomatic, patients complain of joint pain, decreased motion, effusions, and crepitation and, in more advanced cases, may notice deformity due to ongoing bony destruction. Thus, to gather all of these concepts into a broad definition, osteoarthritis should be defined as a degenerative process where continued cartilage breakdown results from mechanical overload, causing secondary bony and synovial changes and characteristic clinical and radiographic findings.
In the hip, there are many new ideas about anatomic and biomechanical factors that may ultimately cause osteoarthritis (OA), and the basic science in this area is evolving rapidly. When evaluating a potential risk factor or cause of a disease, the Bradford-Hill criteria are helpful for determining if an association between a risk factor and a disease is actually a cause-and-effect relationship [3]. These criteria consist of the following:
Strength of association: This refers to the relationship between the possible cause and effect. If there is a stronger relative risk of developing a disease for a patient with a particular risk factor, the risk factor is more likely to be a causal factor. Occasionally, however, the observed association is slight, and the risk factor is nonetheless proven to be a cause of a disease.
Consistency: This means that the same association is observed repeatedly, in studies that occur in different populations, with different study designs, and by different observers.
Specificity: This describes how precisely a potential risk factor can predict that the disease will occur. It is important, however, to keep in mind that diseases may have more than one cause and that one-to-one relationships between a risk factor and a disease are infrequent.
Temporality: This means that the proposed risk factor for the disease always precedes the disease.
Dose–response effect: This means that the frequency of the disease increases with the dose or level of exposure. In orthopedics the dose or level exposure can also be the magnitude of a deformity.
Biological plausibility: This means that, with what is currently known about biology or biomechanics, the proposed risk factor could reasonably cause the disease. Nonetheless, it is important to keep in mind that sometimes the basic science also needs to advance to elucidate the relationship between the cause and effect.
Coherence: This means that the proposed association should not contradict current knowledge about the natural history and biology of the disease.
Experimental evidence: This means that an experiment validates the cause-and-effect relationship in the expected manner. For example, modifying a risk factor decreases the frequency of a disease, or addressing the proposed cause of the disease brings about a cure.
Analogy: This is the process of thinking about a proposed risk factor by comparing other similar and known cause-and-effect relationships for a particular disease. Reasoning by analogy can help to ascertain a cause-and-effect relationship when the observed association is slighter but similar to a known effect.
Hill himself cautioned, however, that these criteria are not necessary or sufficient for making a causal judgment and should be used more as guidelines for considering whether an observed risk factor truly causes a disease [3]. He also reminded the reader that “the ‘cause’ of illness may be immediate and direct, it may be remote and indirect underlying the observed association.” Thus, returning to the question of the etiology of hip OA, it is more likely that the “cause” is multifactorial and different for different individuals. Furthermore, the Bradford-Hill criteria provide a useful framework for evaluating current hypotheses and evidence about the etiology of hip OA.
Although global prevalence of radiographic hip OA varies considerably, it is a common condition in the United States and Europe. The lifetime risk of developing symptomatic hip OA has been estimated to be as high as 25 % after adjusting for race, body mass index, sex, and prior injury [4]. However, not everyone who has radiographic evidence of hip OA becomes symptomatic. In one recent study, only 20 % of people with radiographic hip OA eventually became symptomatic enough to require total hip arthroplasty [5]. The natural history of asymptomatic radiographic hip OA is, however, difficult to elucidate because it requires long-term prospective cohort studies of large populations. Furthermore, the number of patients who progress to arthroplasty is likely to increase because many middle-aged and elderly patients expect to remain active indefinitely and would rather undergo arthroplasty than modify their activities. Age is one of the known risk factors for developing hip OA, and the incidence of hip OA increases with age. Not only does cartilage accumulate damage over time, but older mesenchymal stem cells also have less repair capacity and a decreased ability to protect cartilage from biomechanical stress [6]. Thus, as the expected human lifespan increases, the amount of hip OA and rates of hip arthroplasty are also projected to increase [7].
Other risk factors for hip OA include physical activity like long-term frequent lifting and standing [8] as well as intense or impact sports in young adulthood [9, 10]. There is an association between higher body mass index (BMI) and hip OA, although this association is much weaker than the association between BMI and knee OA [11]. Sex also appears to be a risk factor, with women having higher rates of hip OA than men [4]. Finally, family history and known congenital deformities have also been categorized as risk factors for hip OA.
Historically, hip OA was categorized as primary or idiopathic and secondary, meaning that the hip became arthritic as a result of a prior traumatic injury, pediatric deformity, or following infection. Primary or idiopathic hip OA was attributed to having “bad genes” – essentially that the patient had inherited weak cartilage. However, evidence is accumulating that primary hip OA is actually secondary to a subtle mechanical problem like femoroacetabular impingement (FAI) or mild dysplasia. The concept that hip OA is a mechanical process was proposed by a number of authors and summarized nicely by Ganz in 2008:
Most, if not all, hip OA is secondary, often secondary to subtle but definite and commonly overlooked, ignored, or not recognized dysplasia or pistol grip deformities (FAI). [12]
The Genetics of Hip Osteoarthritis
While there is clearly an inheritance pattern to hip OA, the nature of the genetic contribution is not entirely known. Have family members with a history of hip OA all inherited a bone structure like dysplasia or FAI that causes chondral damage and subsequent OA, or have they simply inherited less rigorous cartilage that is more likely to be damaged in the setting of subtle FAI or dysplasia? Or, as seems likely, is it some combination of the two factors?
Twin studies done in Caucasian females found a genetic contribution of about 60 % for both center-edge angle (as a measure of acetabular depth) and radiographic hip OA [13]. The magnitude of the genetic contribution is not the same for other joints, meaning that the etiology of OA is likely specific to mechanical factors and anatomy at that joint. This also implicates morphology rather than poor-quality cartilage as the bigger risk factor for hip OA. Other studies have shown that femoral head shape is heritable in families with a history of arthroplasty for “idiopathic” OA. However, in one of these studies, patients with a positive family history were more symptomatic than patients with the same degree of FAI morphology but no family history of hip OA. This suggests that bony morphology may not be entirely responsible for symptom development [14]. Genes have been identified that are associated with both cartilage thickness and hip shape [15, 16]. These genes are expressed in developing limb buds and in developing cartilage as well as being expressed in response to increased biomechanical loads [15, 16]. Thus, the genes associated with hip OA could affect either the hip shape or the cartilage microstructure. Finally, genetic variability influences the association between hip OA and bony morphology, meaning that certain genotypes appear to make the cartilage more vulnerable to mechanical overloading from subtle FAI or dysplasia [15].
Acetabular Dysplasia
Acetabular dysplasia is defined as a shallow or small acetabulum that inadequately covers the femoral head. Moderate to severe acetabular dysplasia has long been recognized as a risk factor for the early development of hip OA [17]. The risk of developing hip OA due to mild or borderline dysplasia is less clear, however, and may be influenced by external factors like soft tissue laxity, femoral version, and sport or dance activities.
Although dysplasia has historically been thought of in the context of infantile hip subluxation or dislocation, there is growing recognition that adolescent- or adult-onset dysplasia may represent a developmental process distinct from infantile dysplasia [18]. Furthermore, very few younger adults undergoing hip arthroplasty for arthritis secondary to dysplasia are identified as neonates [19]. The demographics of the infant and adolescent dysplasia populations are different, with adolescent-onset dysplasia patients having more bilateral hip involvement, a stronger family history, and a higher proportion of male patients [18]. Infantile dysplasia may represent a “packaging problem,” meaning that mechanical factors play a greater role in the shape of the neonatal acetabulum and containment of the femoral head. The risk factors for infantile dysplasia – breech positioning, left-sided laterality, and first-born females – implicate the intrauterine environment as a mechanical factor influencing acetabular development. Furthermore, the historical prevalence of dysplasia was substantially higher in populations that had a tradition of infant swaddling with the legs in extension. When this connection was recognized and parents were instructed not to swaddle their children, the incidence of dysplasia decreased [20].
The prevalence of acetabular dysplasia varies widely [20]. It is somewhat difficult to compare the prevalence of dysplasia between countries or regions because some studies have evaluated adults whereas for some populations the data are only available for infants. In addition, some studies have defined dysplasia as a center-edge angle of <25° whereas others have used a center-edge angle of <20°. Nonetheless, it is well recognized that the prevalence of dysplasia is higher in Asia and is the most common cause of hip OA in Japan [21].
A family history of dysplasia is a known risk factor for dysplasia and is consistent across all studied populations. Dysplasia is even more prevalent in areas where consanguinity (e.g., marriage between first cousins) is common [20]. Twin studies have revealed that the heritability of dysplasia is likely polygenic, with a higher incidence of dysplasia in monozygotic twins as compared to dizygotic twins. These findings have led investigators to propose that the genetic mechanism involves inheritance of excessive soft tissue laxity as well as acetabular shape [20].
Biomechanics of Dysplasia
In normal hips, the peak cartilage contact pressure when standing is located near the acetabular dome. The peak contact site varies between the lateral edge and the superior dome of the acetabulum, becoming more medial if the acetabulum is deeper and more lateral if the acetabulum is shallow [22, 23]. There is a direct relationship between the degree of acetabular coverage (as measured using the center-edge angle) and the contact area of the acetabular surface. As the contact surface area decreases, the peak contact pressure increases – meaning that a small center-edge angle is a marker for higher contact pressure [22]. This translates to increased force on the acetabular rim, particularly in stance, and causes characteristic chondrolabral pathology, including labral tears, ganglia, and, in some cases, acetabular rim fractures [24]. The tissue loss predictably occurs at the superior and anterosuperior regions of the acetabulum [24], which corresponds to the area of the highest load [22]. Acetabular version also influences contact pressures. Highly anteverted dysplastic hips have higher anterior contact stresses as a result of minimal anterior femoral head coverage [25]. In contrast, patients with dysplasia and retroversion have impingement-type contact stresses at the anterior edge of the acetabulum. Correcting the version and coverage with an acetabular reorientation osteotomy has been shown to decrease contact pressure by up to 50 % [23].
Natural History of Dysplasia
Radiographic dysplasia, variably defined as a center-edge angle of <20° or <25°, is clearly associated with an increased risk of hip OA [17]. The risk of developing hip OA is also clearly related to the grade of dysplasia, indicating that hips with worse biomechanics and higher contact pressures have a higher likelihood of sustaining joint damage and ultimately becoming arthritic (Fig. 2) [17]. If hip pain in a young person (<40) is considered to be a precursor of hip OA, it is notable that 25–35 % of young patients with hip pain have dysplasia [26]. Version may also play a role in the natural history of dysplasia. Patients who have retroversion and dysplasia experience an earlier onset of hip pain as compared to patients with normal anteversion [27].
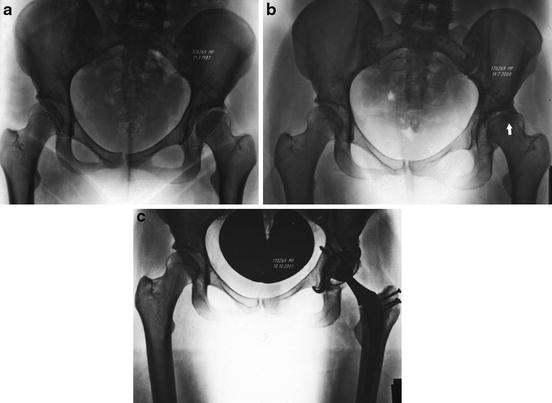
Fig. 2
The natural history of a patient with severe left anterior dysplasia (outline). At the time of presentation, the hip had already subluxed laterally, but the joint space was relatively preserved (a). Thirteen years later, there was advanced and severe cartilage degeneration on the femur, with a subchondral cyst and sclerosis (arrow) in the femoral head, as well as on the acetabulum (b). She underwent a total hip arthroplasty 1 year later (c)
If the loading biomechanics of a dysplastic hip are changed as a result of a femoral or acetabular osteotomy, the natural history of that hip appears to improve. The results are better for periacetabular or rotational acetabular osteotomy than for femoral osteotomy however. The long-term (20-year) survival rate of the native hip after a periacetabular osteotomy is about 60 % [28]. Even with the improvement in hip biomechanics, most patients have some progression of osteoarthritis and, on average, advance one radiographic Tönnis grade after 10 years [28]. Because dysplasia is largely a problem related to static loading across the hip, one might expect that weight loss in an overweight patient with dysplasia could improve hip pain and natural history because it decreases the overall static load. Although weight loss is known to improve pain and function in patients with knee OA, this has not been studied for patients with dysplasia. The potential for an osteotomy to improve hip function does have some limits. The success rates of osteotomy are poor for patients older than 35 with Tönnis grade 2 or more radiographic hip OA [28]. Thus, there appears to be a “tipping point” of cartilage damage, after which an osteotomy is unlikely to improve the natural history of a dysplastic hip.
Femoroacetabular Impingement (FAI)
Broadly speaking, FAI is defined as the abnormal contact between the femur and the acetabulum during hip motion that occurs as a result of a subtle deformity at the femoral head-neck junction or acetabular rim and that causes progressive chondrolabral damage [29]. Although Reinhold Ganz generally receives credit for describing FAI [29], the earliest description of impingement appears to have been in 1899 in the French literature, with the author noting “empreinte iliaque,” or an impression on the head-neck junction produced by the ilium at the area of the anterior-inferior iliac spine with the hip in flexion [30]. Subsequent authors correctly described impingement in the context of coxa vara, severe protrusio deformities, and slipped capital femoral epiphysis [31–33]. Depending on the site of the deformity, these authors also recommended femoral neck osteoplasty and/or acetabular rim trimming to restore range of motion and provide pain relief. In contrast to all of the previous authors however, Ganz substantiated his ideas about FAI with observations of chondrolabral damage at the site of the impinging lesions and with the results of treatment [29, 34], both of which were made possible with the development of the technique for a safe surgical dislocation of the hip. The description of FAI also coincided with technical improvements in hip arthroscopy that resulted in an increase in hip arthroscopy for labral tears. As a result, arthroscopists began to recognize and describe early chondrolabral damage, which ultimately helped to substantiate the association between hip pain, impingement anatomy, and eventual hip OA [35].
Related posts:
 Hip Arthroscopy: Supine Approach to Patient Positioning, Set-up, and Traction
Hip Arthroscopy: Supine Approach to Patient Positioning, Set-up, and Traction
 Surgical Technique: Bone Graft for Avascular Necrosis of the Hip
Surgical Technique: Bone Graft for Avascular Necrosis of the Hip
 Rehabilitation of Non-Operative Hip Conditions
Rehabilitation of Non-Operative Hip Conditions
 Surgical Technique: Open Proximal Hamstring Repair
Surgical Technique: Open Proximal Hamstring Repair
 Subspine Impingement and Surgical Technique
Subspine Impingement and Surgical Technique
 Atraumatic Instability and Surgical Technique
Atraumatic Instability and Surgical Technique
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


