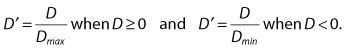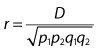21 Genetics of Rheumatic Diseases
HLA molecules are among the most highly variable (polymorphic) proteins encoded in the genome.
HLA variability is a major factor in controlling immune responses.
Many of the newly defined risk alleles predispose to multiple different autoimmune disorders.
A large fraction of the genetic risk for autoimmunity remains to be defined.
Major Histocompatibility Complex
The MHC, which encodes the human leukocyte antigens (HLAs), has been associated with susceptibility to many different diseases since the late 1970s. The chromosomal region containing the MHC was originally identified because of the ability of genes in this region to regulate transplant rejection1 and to control the immune responses of mice and guinea pigs to simple antigens,2 a series of observations that led to the 1980 Nobel prize. The HLA molecules and their counterparts in rodents were subsequently shown to be directly responsible for immune response differences between individuals and for determining the likelihood of graft rejection.1–4 A large number of different HLA genes exist within the MHC, and they exhibit an enormous degree of structural variability. In addition to influencing immune response patterns, many of these alleles are associated with susceptibility to a wide spectrum of autoimmune diseases, making the MHC an essential starting point for anyone wanting to understand the genetics of rheumatic diseases.
Human Leukocyte Antigen Molecules and Antigen-Specific T Cell Recognition
The primary function of HLA molecules is the presentation of antigenic peptides to T cells. In the case of α/β T cells, most antigen recognition events involve the formation of a trimolecular complex consisting of the HLA molecule, its bound peptide, and the α/β T cell receptor (see Chapters 13 and 19). When this recognition occurs in the appropriate context, it may result in signal transduction and activation of the T cell. The requirement for MHC molecules to present antigenic peptides to T cells is frequently referred to as “MHC-restricted” T cell recognition. In each individual, T cells are generally restricted to recognize antigens presented by the person’s own HLA molecules. The allelic variations among different HLA molecules are a major factor accounting for differences in the types of antigenic peptides to which an individual responds or in the types of T cells that are used in an immune response.
Human Leukocyte Antigen Class I and Class II Molecules
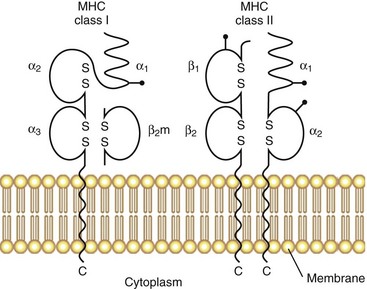
The original serologic and biochemical studies of HLA molecules revealed the presence of two major isotypes: HLA class I and HLA class II. The basic structural features of these classical HLA molecules are summarized in Figure 21-1. HLA class I molecules consist of a 45-kD α chain encoded within the MHC that is noncovalently associated with the 12-kD β2-microglobulin chain (encoded on chromosome 15). HLA class II molecules consist of noncovalently associated α (32 kD) and β (28 kD) chains, both of which are encoded within the MHC. HLA class I and class II molecules are cell surface glycoproteins, anchored to the membrane by hydrophobic transmembrane segments.
A major breakthrough in the understanding of HLA molecules came in 1987, when Bjorkman and colleagues5,6 reported the crystal structure of the HLA class I molecule, HLA-A2. This work was followed by the solution of other MHC class I and class II structures relevant to rheumatic disease.7,8 A side view of the class I molecule taken from Bjorkman’s original paper is shown in Figure 21-2A. It can be seen that the base of the molecule (directly adjacent to the cell membrane) is formed by β2-microglobulin and the immunoglobulin-like α3 domain. The α1 and α2 domains form a distinct cleft or groove at the top of the molecule. The function of this cleft is to bind antigenic peptides for presentation to T cells. A top view of this cleft is shown in Figure 21-2B. One can appreciate that the “floor” of the peptide binding cleft consists of β-sheets, whereas the “walls” of the cleft are bounded by extended regions of α-helical structure. The size of the HLA class I cleft is approximately 10 to 20 angstroms and generally can accommodate antigenic peptides that are 9 amino acids long.9,10
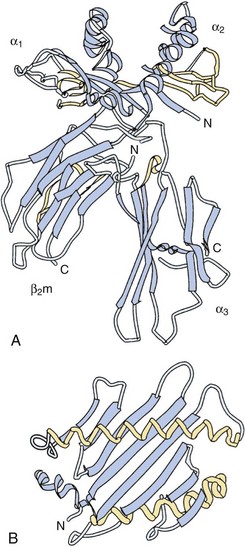
Figure 21-2 Three-dimensional structure of an HLA class I molecule, based on the X crystallographic analysis.5 A, Side view. A peptide-binding cleft is formed by the α1 and α2 domains at the top of the molecule. The α3 and β2 microglobulin (β2m) domains are similar in structure to immunoglobulin domains; essentially, they act as a platform on which the peptide binding cleft rests, as well as providing contact sites for the CD8 molecule during CD8+ T cell recognition. B, Top view of the empty peptide-binding cleft. This “T cell view” of the MHC molecule would normally include a peptide bound within the cleft. Note the disulfide bond, which connects the α-helix of the α2 domain with the floor of the cleft.
HLA class II molecules have a structure that is highly similar to that of class I molecules, with a prominent peptide binding cleft at the membrane distal portion, sitting on top of a base formed by the α2 and β2 immunoglobulin-like domains. The overall size and shape of the cleft in the two classes of HLA molecules are almost superimposable. However, subtle differences exist, particularly at the ends of the cleft, and this allows for some differences in the sizes of antigenic peptides that are presented by HLA class II molecules, compared with class I. Direct analysis of peptides bound to HLA class II molecules has shown that their size commonly varies from 12 to 19 amino acids.10 Thus relatively longer peptides lie in the cleft of class II molecules and may extend beyond the ends of the cleft, whereas class I molecules contain much shorter peptides that are buried within the cleft at either end.
Finally, x-ray crystallographic analyses of the entire trimolecular complex consisting of an HLA molecule, its bound peptide, and the T cell receptor exist. Figure 21-3 shows an example of this structure for an HLA-DRB1*0401 allele presenting influenza Ha Peptide to its cognate α/β T cell receptor.11 The Protein Data Bank (PDB) website (www.rcsb.org/pdb/home/home.do) provides three-dimensional versions of this HLA structure (Entry code 1J8H).
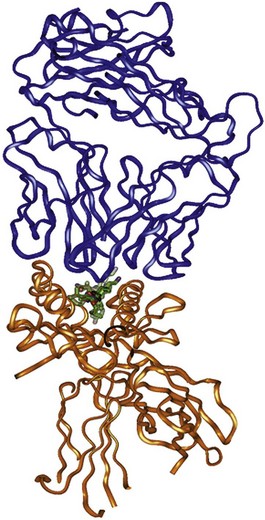
Figure 21-3 Ribbon diagram derived from the three-dimensional crystal structure of a the trimolecular complex of a human α/β T cell receptor (top, green), influenza Ha antigen peptide, and the MHC class II molecule, DRB1*0401.11 Note that the peptide is contained with the peptide-binding cleft of the HLA-DR molecule. The polymorphisms associated with the “shared epitope” are located on the α-helical rim (DRB1 chain) of the peptide-binding cleft, where they may interact with either the bound peptide antigen or the T cell receptor.
Human Leukocyte Antigen Class I and Class II Isotypes: Functional Correlates
Another major functional difference between class I and class II molecules is related to the source of peptide antigens that are found in the antigen-binding cleft. In general, class I molecules present peptide antigens derived from proteins that are actively synthesized within the endoplasmic reticulum, whereas HLA class II molecules present antigens that are taken up from outside of the cell by endocytosis. These differences are reflected in the antigen processing machinery and the different trafficking patterns of class I and class II molecules inside the cell. Chapters 10 and 19 discusses this complex process in detail.
Genetic Organization of the Human Major Histocompatibility Complex
The human MHC extends over approximately 4 million base pairs on the short arm of chromosome 6 (6p21.3). The HLA class I and class II gene clusters are found in distinct locations, as indicated on the highly abridged genetic map shown in Figure 21-4. Only those genes that are traditionally associated with immune function are shown in Figure 21-4. The MHC, one of the most gene-rich regions in the human genome, has identified more than 200 genes.12
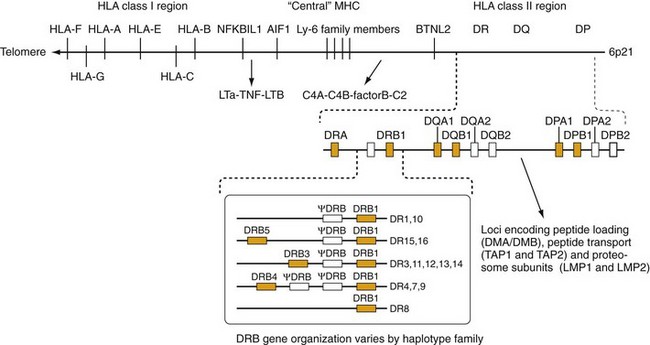
Figure 21-4 Map of the human major histocompatibility complex (MHC). The HLA class I and class II molecules are encoded in distinct regions of the MHC. The HLA class II region contains three subregions: DR, DQ, and DP. Each of these contains a variable number of α- and β-chain genes. HLA class II loci with known functional protein products are labeled in bold. In the case of DR, different numbers of DRB genes are present in different haplotypes. A summary of the most common of these is shown in the box. The DQ and DP subregions each contain one pair of functional α- and β-chain genes. A number of genes involved in antigen processing and presentation by class I molecules are situated between the DP and DQ subregions. The HLA class I region contains the three “classical” class I genes, HLA-A, HLA-B, and HLA-C, as well as other related class I molecules (see text). The “central MHC” also contains a number of genes related to immune function including the complement components (C4A, C4B, C2, and factor B), as well as tumor necrosis factor and lymphotoxin. Other potentially interesting genes in this region (NFKB1L1, AIF1, and BTNL2) are discussed in the text and reference 68.
The HLA class I α-chain genes are on the telomeric side of the MHC including the classical class I genes, HLA-A, HLA-B, and HLA-C; these three genes are also referred to as the class Ia genes. Researchers have defined several other class I genes including the HLA-G, HLA-E, and HLA-F loci, also known as class Ib genes. Initially, experts considered these class Ib genes to be nonfunctional because they exhibit limited polymorphism. However, some of these genes clearly have significant immune functions. For example, HLA-E functions as a ligand for natural killer (NK) cell receptors.13 In a surprising twist, the cell surface expression of HLA-E molecules often depends on binding peptides derived from the signal sequence of class I molecules,14 but they can also present antigenic peptides to α/β TCR-bearing CD8+ cells.15 Despite its limited polymorphism, alleles of HLA-E may still affect responses to some viral infections.15,16
The HLA class II genes are situated centromeric to the class I region and have a somewhat more complicated organization. The three major subregions of the class II cluster are designated DR, DQ, and DP. Each of these subregions contains a variable number of α- and β-chain genes. Particularly in the case of the DR subregion, this variability has led to confusion regarding the nomenclature to describe these genes. Experts have agreed on an international standard for this nomenclature regularly update it (see www.ebi.ac.uk/imgt/hla/).
The DR subregion contains a single α-chain gene, designated DRA, which does not exhibit significant allelic variation. In contrast, the genes encoding the DRβ chains (DRB) are highly polymorphic and vary in number among different individuals in the population. The boxed area in Figure 21-4 shows several examples of common DR haplotypes. (A haplotype refers to a group of alleles at closely linked loci that are commonly inherited together.) Many of these DRB genes are nonfunctional pseudogenes (indicated by the symbol ψ), although all haplotypes contain at least one functional DRB1 gene, and many haplotypes contain a second functional DRB gene (DRB3, DRB4, or DRB5).
In addition to the HLA class II molecules, several other genes distributed within the class II region are involved in peptide antigen processing. The TAP1 and TAP2 genes have been of particular interest because they exhibit a modest degree of polymorphism and are involved in delivering peptides for loading onto HLA class I molecules.17 Other proteins encoded in this region are involved in peptide loading onto class II molecules18 such as the DM molecule (encoded by the DMA and DMB genes) and the DO/DN heterodimer (encoded by DOB and DNA).
Human Leukocyte Antigen Molecules Are Highly Polymorphic
One of the most dramatic features of the HLA system is the extreme degree of polymorphism at most of these loci. The formal definition of polymorphism (Table 21-1) usually requires that the most common allele at the locus does not exceed a frequency of 98%. In contrast, at many HLA loci, it is uncommon for a single HLA allele to exceed a frequency of 50% in the population. The number of different alleles present in the population is much larger than in any other known polymorphic locus encoding functional genes. For example, at the HLA-A locus, more than 100 different alleles have been reported; at HLA-B, the number of reported alleles exceeds 200. A similar degree of allelic diversity is seen at the DRB1 locus and to a lesser degree at DQA and DQB.
| Allele | Alternative form, or variant, of a gene at a particular locus |
| Alloantisera | Antisera that detect antigenic differences between individuals in the population; the term is most often used to refer to sera that detect antigenic (i.e., structural) differences among human leukocyte antigen molecules carried by different individuals |
| Haplotype | A group of alleles at adjacent or closely linked loci on the same chromosome that are usually inherited together as a unit |
| Heterozygote | An individual who inherits two different alleles at a given locus on two homologous chromosomes |
| Heterozygosity | A measure at a particular locus of the frequency with which heterozygotes occur in the population |
| Linkage | The tendency toward the co-inheritance within a family of two genes that lie near each other on the genome; complete linkage occurs when parents who are heterozygous at each locus are unable to produce recombinant gametes |
| Linkage disequilibrium | The preferential association in a population of two alleles or mutations that occurs more frequently than predicted by chance; linkage disequilibrium is detected statistically, and except in unusual circumstances, it implies that the two alleles lie near each other on the genome |
| Polymorphism | The degree of allelic variation at a locus within a population; specific criteria differ, but a locus is said to be polymorphic if the most frequent allele does not occur in > 98% of the population; occasionally, polymorphism can be used in the same way as allele to refer to a particular genetic variant |
| Penetrance | The conditional probability of disease (or phenotype) given the presence of a risk genotype |
For many years the naming of these various HLA alleles was a major source of confusion in the literature. The difficulty with the nomenclature stems in part from the different methods that have been used to define HLA polymorphisms. Originally, HLA class I alleles were detected through the use of alloantisera (see Table 21-1); the prefix “allo-” refers to genetic differences that exist between individuals of the same species. Alloantisera directed toward HLA molecules are commonly found in the context of pregnancy, in which the mother mounts an immune response against the “foreign” HLA molecules carried by the fetus (derived from the father). Anti-HLA responses are also seen after blood transfusion because the HLA molecules on the donor cells are highly immunogenic. In the case of HLA class II alleles, differences were originally detected using mixed lymphocyte responses. When T cells from a responder are mixed with lymphocytes from another individual, differences in HLA class II alleles cause the responder’s T cells to proliferate. Data on mixed lymphocyte culture (MLC) typing dominated the early HLA literature, and it was the method first used to detect the HLA class II associations with RA.19 Subsequently, serologic methods were also employed to detect class II polymorphisms.
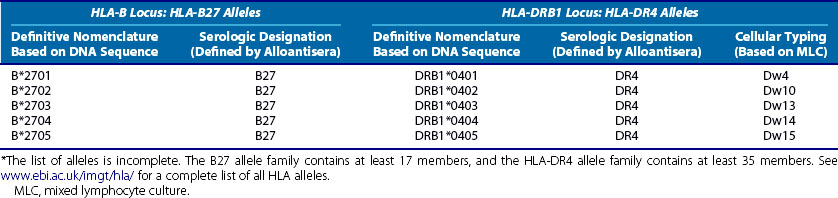
The current names of the HLA class I and class II alleles are attached to the specific DNA sequence and locus for each allele and are definitive. However, many older publications have used the serologically derived names for alleles. It is therefore important to have some concept of how these naming conventions are related. The modern definitive (sequence-based) allele names are derived from the older serologic names because the serologic techniques frequently detected whole groups of related alleles. Two examples of this are shown in Table 21-2. The designation of HLA-B27 was developed for people carrying an HLA-B allele that was recognized by the B27-specific alloantisera. However, sequencing of the HLA-B alleles carried by these individuals revealed the existence of at least 17 different alleles, 5 of which are listed in Table 21-2. A similar situation exists for HLA class II allele families such as HLA-DR4, also shown in Table 21-2. In this case, the DR4 allospecificity was already known to detect a number of different alleles that could be further discriminated on the basis of MLC typing.20 These have been defined and named by their sequence, as shown in Table 21-2. A full list of all HLA alleles at the major loci can be found at www.ebi.ac.uk/imgt/hla/.
Table 21-2 Comparison among Modern, Sequence-Based Nomenclature, and Older Naming Conventions for Class I and Class II Alleles Belonging to HLA-B27 and HLA-DR4 Serologic Groups*
Human Leukocyte Antigen Associations with Rheumatic Diseases
Population-Association Studies and the Calculation of the Odds Ratio, an Estimate of Relative Risk
The ideal way to establish whether a genetic variant (allele) confers risk for a disease is by performing a prospective cohort study. In this kind of study, a group of individuals carrying (exposed to) the allele is compared with a matched control group that does not carry the allele. These two groups are followed over time (preferably over a lifetime) to see if disease develops more frequently in the exposed group. The results can be displayed in a contingency table (Table 21-3). By examining the upper half of Table 21-3, it is apparent that the fraction of exposed individuals who get the disease is a/(a + b), whereas the fraction of unexposed individuals who develop the disease is c/(c + d). The ratio of these two fractions is known as the relative risk (RR) = a/(a + b) ÷ c/(c + d) = (ac + ad)/(ac + bc). If the disease is rare in the population, ac is small and the RR is approximated by (a × d)/(b × c), also referred to as the cross-product.
Table 21-3 Contingency Table for Cohort and Case-Control Studies*
| Cohort Study | ||
| Disease | No Disease | |
| Exposed | a | b |
| Not Exposed | c | d |
| Case-Control Study | ||
| Exposed | Not Exposed | |
| Disease | a | b |
| No Disease | c | d |
* a, b, c, d, number of individuals observed in each category.
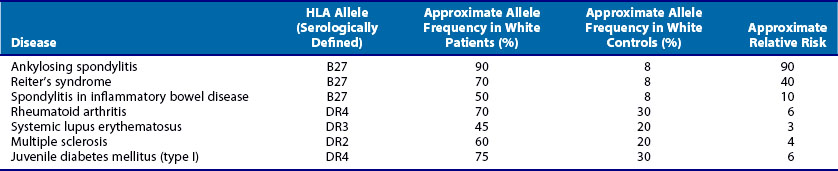
In reality, such prospective cohort studies are usually impractical and therefore a retrospective case-control design is used. In this type of study, subjects are initially identified according to whether they have the disease and individuals without the disease are the controls. The data can be tabulated as in the lower half of Table 21-3. In this case, the cross-product or (a × d)/(b × c) is known as the odds ratio (OR). In practice, this quantity is often reported as the estimated RR because the cross-product is close to the RR when the disease is rare. An OR of 1 indicates that the genetic factor confers no risk for the disease. An OR less than 1 suggests that the genetic factor under study is negatively associated with the disease. (ORs of less than 1 are occasionally reported as the negative inverse value; an OR of +0.5 may also be reported −2.0.) With the exception of HLA-B27–associated diseases, most HLA associations with rheumatic diseases have ORs of less than 10. Several examples of typical HLA associations with rheumatic and autoimmune disorders are shown in Table 21-4.
Human Leukocyte Antigen Class I Associations: HLA-B27 and Spondyloarthropathies
One of the strongest and earliest21 reported HLA associations with the rheumatic diseases is the association of HLA-B27 with ankylosing spondylitis (AS). In white populations, more than 90% of patients with AS carry HLA-B27, in contrast to approximately 8% of normal individuals, giving estimated RR values of 50 to 100 or higher. The consistency of this finding across most ethnic groups lends support to the contention that the HLA-B27 alleles are directly involved in the pathogenesis of AS.22,23 HLA-B27 is also associated with reactive arthritis and with the arthritis seen in the context of inflammatory bowel disease. As shown in Table 21-4, the strength of these associations is lower in terms of estimated RR compared with ankylosing spondylitis.
The serologic specificity of HLA-B27 actually encompasses many distinct HLA class I alleles. These alleles differ from one another at a number of amino acid positions, most of which involve amino acid substitutions in and around the peptide binding pocket. This fact leads naturally to the question of whether there are differences among these B27 alleles in terms of disease association. Most data indicate that this is not the case, although there may be some exceptions in some populations.22 These exceptions may provide clues to the role of the HLA-B27 molecule in pathogenesis. Overall, however, it appears that most of the structural differences among the B27 alleles do not affect disease risk.23
In recent years it has become apparent that increased risk for some severe drug reactions can be ascribed to HLA24 including risk for allopurinol-associated Stevens-Johnson syndrome in Asian populations with HLA-B58.25 These data imply the need for specific class I HLA typing before treatment with some medications.26
Human Leukocyte Antigen Class II Associations with Autoimmune Diseases
A large number of HLA class II associations with autoimmune diseases were described over 2 decades ago.27 RA has received particularly intense scrutiny over the years, but the precise reasons for the HLA associations with this disease are still unknown. In the case of systemic lupus erythematosus (SLE) and related illnesses, many of the HLA class II alleles are associated with the presence of specific autoantibodies or clinical phenotypes. Interestingly, the recent data in RA indicate that the major HLA-DR associations are with anti-CCP antibody positive disease, suggesting that control of autoantibody responses may be a primary mechanism underlying these associations in RA as well.
Rheumatoid Arthritis: HLA-DRB1 Associations and the “Shared Epitope”
Stastny19 reported the first associations of rheumatoid arthritis (RA) with HLA class II alleles in the 1970s. This was done using cellular28 and antibody reagents29 that are no longer routinely used for HLA typing; however, as discussed earlier, the nomenclature for HLA alleles still derives from these early typing methods. The DRB1*0401 allele (corresponding to the “Dw4” type in Stastny’s original report28) was the first HLA polymorphism to be associated with RA. Numerous studies have generally confirmed that this allele is the most strongly associated with RA, at least in white populations.30–32 However, several other HLA-DRB1 alleles have also been associated with RA, although the strength of these associations varies.31,33,34 In some ethnic groups, RA is not associated with HLA-DR4 alleles, but rather with HLA-DR135 or HLA-DR10.36 Experts now widely accept that the following alleles are the major contributors to RA risk at the DRB1 locus: DRB1*0401, -0404, -0405, -0101, and -1001. In addition, minor variants of these alleles and others (e.g., DRB1*140237) may also contribute to susceptibility and DRB1*0901 is a susceptibility allele in Asians, where this allele is common.38 Most of these risk alleles share a common sequence, as shown in Table 21-5. This consensus amino acid sequence 70Q or K-R-R-A-A74 has been termed the shared epitope.39 This structural feature is located on the α-helical portion of the DRβ chain in a position where it may influence both peptide binding and T cell receptor interactions with the DRB1 molecule. (In the case of the DRB1*1001 risk allele, one amino acid varies from this consensus by a conservative change, with an R at position 70, as does DRB1*0901, which is commonly associated with RA in Asian populations) (Table 21-6).
Table 21-5 Amino Acid Substitutions That Compose the Shared Epitope at Positions 70 through 74 of DRB1 Alleles Associated with Rheumatoid Arthritis
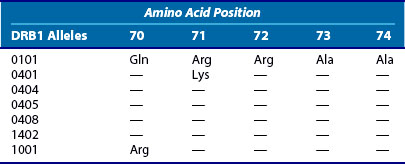
Table 21-6 Genotype Relative Risks of DRB1 Genotypes for Rheumatoid Arthritis
| DRB1 Genotype | Relative Risk | P Value |
|---|---|---|
| 0101/DRX | 2.3 | 10−3 |
| 0401/DRX | 4.7 | 10−12 |
| 0404/DRX | 5 | 10−9 |
| 0101/0401 | 6.4 | 10−4 |
| 0401/0404 | 31.3 | 10−33 |
From Hall FC, Weeks DE, Camilleri JP, et al: Influence of the HLA-DRB1 locus on susceptibility and severity in rheumatoid arthritis, Q J Med 89:821-829, 1996.
A number of different hypotheses have been advanced to explain the shared epitope association with RA.40,41 Two of these follow directly from knowledge about the role of HLA molecules in antigen presentation and immune regulation. Thus it has been suggested that a particular peptide antigen, or set of related antigens, may be involved in the initiation or propagation of RA, and that shared epitope positive DRB1 alleles possess a unique, or enhanced, ability to bind or present these peptides to the immune system.40 It has been difficult to address this hypothesis directly because the identity of these putative disease-causing peptide antigens is unknown. In view of the strong association of the shared epitope alleles with anti-CCP antibodies,42 it is of interest that citrullinated peptides may have a particular affinity for DRB1*0401 alleles.43 A second major hypothesis posits that these risk alleles regulate the formation of the peripheral T cell repertoire, by acting to select for particular T cell receptors during thymic selection.41 There is elegant experimental evidence in humans to support a role for DR4 alleles in shaping the peripheral T cell repertoire.44 However, it is unclear whether this effect on the TCR repertoire is really related to disease susceptibility. Researchers have proposed a number of other interesting hypotheses, involving molecular mimicry,45,46 allele specific differences in intracellular trafficking,47 and regulation of nitric oxide production,48 but these require further experimental confirmation.
The shared epitope hypothesis has come under scrutiny, with some investigators proposing a direct role for HLA-DQ polymorphisms,49,50 in part based on studies in transgenic mice.51 As can be seen in Figure 21-4, the HLA DQ α and β chains are encoded just centromeric to DRB1, and alleles at this locus are in strong linkage disequilibrium with DRB1 alleles. The strong linkage disequilibrium between the DR and DQ loci makes it difficult to tease apart the effects of DR versus DQ solely on the basis of population genetic studies; the arguments for a DQ effect generally depend on showing the enrichment of relatively rare genotypes in the RA patient group compared with controls. Overall, a primary role for DQ alleles is not strongly supported by large HLA association studies that have examined this issue52 and is not supported by more recent dense single-nucleotide polymorphism (SNP) mapping efforts within the MHC.53 Rather, a possible additional role for the HLA-DP has been suggested.53
Regardless of whether DQ or DP alleles are involved in RA susceptibility, it is quite clear that the shared epitope hypothesis is not a complete explanation for the HLA associations with RA. This is evident from the fact that not all SE-positive alleles carry the same degree of genetic risk and the strength of the association varies in different populations. In general, DRB1*0101 alleles carry lower levels of RR for RA than the DRB1*0401 and 0404 alleles,32 and yet DRB1*0101 is the major risk allele in some ethnic groups.54,55 The shared epitope itself does not appear to associate strongly with RA in African-American and some Hispanic populations.56,57 Furthermore, certain combinations of DRB1 alleles carry especially high risk, as originally observed by Nepom.58 Thus the combination of DRB1*0401 with *0404 carries a RR of higher than 30 in Caucasian populations.32 This compares with RR values in the range of 4 or 5 for either allele alone. Table 21-5 summarizes some of these relationships. Recently, attempts have been made to formalize the gradient of risk conferred by the various shared epitope alleles.59 However, it remains unclear whether these effects are mediated by the HLA-DR molecules themselves or reflect the action of other genes on these haplotypes.
HLA-DQ Associations with Autoimmune Diseases
Many of the first HLA class II associations with autoimmune disorders were detected using alloantisera for HLA-DR alleles, as indicated in Table 21-4. However, as knowledge increased about the genetic organization of the class II region, it became apparent that for some diseases, the genetic associations are stronger with HLA-DQ alleles. For example, although juvenile diabetes does exhibit HLA associations with both HLA-DR4 and HLA-DR3, it is likely that a group of associated HLA-DQ alleles actually are responsible for these observations.60 As discussed later, the HLA associations with particular autoantibodies in systemic lupus also probably reflect the effects of HLA-DQ alleles.
The DQ subregion presents special challenges for the newcomer to HLA because the old serologic nomenclature does not usually have a simple correlation with a group of alleles at a single locus. Because most of the HLA correlations with autoantibodies in lupus involve the DQ loci, it is important to understand this at the outset. The problem arises because both the α and β chains are polymorphic in DQ molecules. The serologic specificity of DQ2 may detect one of three closely related DQB1 alleles: DQB1*0201, DQB1*0202, or DQB1*0203. This is analogous to the DR serologic specificity detecting a group of related DRB1 alleles (see Table 21-2). However, in the case of DQ, the DQ2 serologic specificity also detects these alleles on several different haplotypes that may encode quite different DQ α chains. (This is different from HLA-DR molecules, in which the DR α-chain structure is constant and does not vary between haplotypes.) In white populations, the DQB1*0201 allele is commonly found on DR3 haplotypes (associated with DQA1*0501) and DR7 haplotypes (associated with DQA1*0201), but both these haplotypes would type serologically as DQ2 (Figure 21-5). Especially when reading the older literature and discussing DQ polymorphisms, it is important to distinguish serologically defined polymorphisms, which may vary within the group of alleles on the α and β chains, from polymorphisms defined by sequence at a specific locus (DQA1 or DQB1).
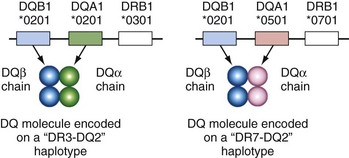
The HLA associations with the Ro (SS-A) and La (SS-B) autoantibody systems have been thoroughly studied. The anti-Ro response is present in 25% to 50% of patients with lupus61 and even more frequently in the setting of primary Sjogren’s syndrome.62 Although early serologic studies indicated an association with HLA-DR3 and DR2, a detailed molecular analysis of these HLA haplotypes has provided evidence that HLA-DQ alleles in linkage disequilibrium with DR2 and DR3 are responsible for controlling this autoantibody response; heterozygous individuals who inherit a DR2-DQ1 haplotype and a DR3-DQ2 haplotype tend to have high anti-Ro antibody titers in the setting of lupus or Sjogren’s syndrome.63 The strongest associations involve a DQA1*0501-DQB1*0201 haplotype (frequently found in linkage disequilibrium with DR3) and a DQA1*06-DQB1*06 haplotype (frequently found in linkage disequilibrium with DR2). HLA-DQ associations have also been reported for other autoantibody systems such as antiphospholipid antibodies64 and anti-Sm responses.65 The overall pattern of HLA-DQ associations with these antibody responses is similar to those seen for anti-Ro responses, although the alleles involved are quite different.
Population-Association Studies: What Do They Mean?
First, the allele under investigation may be directly involved in the pathogenesis of the disease. This assumption actually underlies most of the foregoing discussion on HLA and rheumatic disease. The studies described reflect the search for a more precise definition of particular amino acid substitutions or unifying structural characteristics of disease-associated alleles. This effort derives from the idea that HLA alleles directly predispose to disease by virtue of their ability to control the immune response.4 As discussed earlier, this may involve a number of mechanisms including preferential peptide binding and the influence of MHC on thymic selection of the peripheral T cell repertoire.
A second reason that must be addressed with any new genetic association is the possibility that the result is an artifact of population stratification of patients and controls. The specific concern is that the control group may not be genetically matched to the disease group at loci that are unrelated to disease. This often results from a failure to study a control group that is ethnically matched to the disease group. This is a major issue generally in genetic case-control studies, and several approaches to control for this have been proposed66 including the use of panels of genetic markers that specifically reflect ethnic background.67 These methodologies for correcting for underlying population are now widely accepted and indeed are often required for publication in leading genetics journals. It is generally not adequate to accept self-reported ethnicity as a basis for matching cases and controls.
Finally, a third (and common) reason for observing a genetic association is that the causative gene is actually in linkage disequilibrium with the marker allele being tested, be it a SNP or a particular HLA variant. Linkage disequilibrium is discussed in greater detail in the next section and refers to the fact that genetic variants at adjacent loci often tend to be found together more frequently than expected by chance. Linkage disequilibrium over long distances is a particularly prominent feature of the HLA region, particularly for certain haploytpes.68,69 A good example of how HLA associations can reflect linkage disequilibrium with a gene that is functionally unrelated to HLA is hemochromatosis. Early studies showed that certain HLA class I alleles such as HLA-A3 were highly associated with this disorder. However, it is now clear that the causative gene, HFE, is actually more than 3 million base pairs distant from the HLA-A locus (toward the telomere in Figure 21-4). The HLA-A3 association is observed simply because the HFE C282Y allele (causative for hemochromatosis) is frequently found on the same haplotype (see Table 21-1) as the HLA-A3 allele in many white populations.
Because there are a number of genes with immunologic function within the MHC complex that may themselves be directly involved in predisposing to autoimmunity, these loci must always be considered as possible causative genes when considering the significance of a new HLA association with rheumatic disease. Indeed, recent studies of autoimmune diseases indicate that multiple genes within the MHC can contribute independently to disease risk. In the case of RA, a number of studies have indicated that a separate locus in the central MHC may associate with the disease, independently of the HLA-DRB1 locus.70–72 In addition, there is evidence that genes in the class I region may influence the risk conferred by certain HLA-DRB1*0404 haplotypes72 or may interact with other non-MHC genes.73 Similar analyses in type 1 diabetes,74 lupus75,76 juvenile arthritis,77,78 multiple sclerosis,79 and myasthenia80 all point to the fact that multiple different genes within the MHC can contribute to disease susceptibility. This issue is currently a major focus of research efforts in autoimmune diseases, and it is highly likely that additional risk genes in the MHC will be defined in the next few years using the dense SNP mapping techniques discussed in the following sections.
Linkage Disequilibrium
The concept of linkage disequilibrium is central to understanding the significance of any genetic association including HLA associations with disease. Linkage disequilibrium exists when the frequency of two alleles occurring together on the same haplotype exceeds that predicted by chance. For example, a common MHC haplotype that exhibits linkage disequilibrium in the white population carries a certain combination of alleles, A*0101-B*0801-DRB1*03011, commonly referred to as the A1-B8-DR3 haplotype, and more recently designated the “8.1” haplotype.68 This haplotype is present in about 9% of the Danish population, a typical white Northern European group. To understand why this reflects the presence of linkage disequilibrium, consider the fact that the A1 allele is present in 17% of Danes and the B8 allele is present in 12.7% of Danes. They could be expected to be found together only 12.7% × 17% = 2.1% of the time, much less than what is observed (9%). This simple difference between the expected and observed association between alleles is a measure of linkage disequilibrium, known as D; in this case D = (0.09 − 0.021) = 0.069. Table 21-7 provides the general calculation of D for a simple two-locus, two-allele situation. Because the magnitude of D is strongly influenced by the relative frequencies of the various alleles, normalized measures of linkage disequilibrium are used in practice including D′ and r2, as described in Table 21-7.
Table 21-7 Measuring Linkage Disequilibrium
| Consider a region of a chromosome with two adjacent loci, A and B. At each locus, there are two possible alleles, 1 and 2. There are four possible combinations of alleles, or haplotypes. These are shown below including a color bar representation and designations for the frequency of these haplotypes in the population. |
A1____B1  frequency = x11 frequency = x11 |
A1____B2  frequency = x12 frequency = x12 |
A2____B1  frequency = x21 frequency = x21 |
A2____B2  frequency = x22 frequency = x22 |
| Designate the allele frequencies at locus A as p1 and p2. |
| Designate the allele frequencies at locus B as q1 and q2. |
| Then |
| p1 = x11 + x12 |
| p2 = x21 + x22 |
| q1 = x11 + x21 |
| q2 = x12 + x22 |
| A simple measure of D is then calculated as D = x11− p1* q1. This value directly measures the difference between the observed haplotype frequency (in this case x11) from that expected from random association of the alleles at each locus (in this case p1* q1). For any allelic combination in a given dataset, the magnitude of D will be the same, although the sign (+ or −) may change according to the direction of the allelic association on the haplotypes. |
| A more standardized measure of D is more useful in practice and is designated D′. D′ is the ratio of the observed D to the maximal (or minimal) possible value of D given the observed allele frequencies. |
|
| D′ can therefore take on values between 0 and 1, and it is a measure of linkage disequilibrium that is normalized for variations in allele frequencies at the two loci. |
| Another useful measure of LD is the correlation coefficient, r2, between alleles at A and B. Like D′, the value of r2 can also vary between 0 and 1. However, unlike D′, the value of r2 is a more global measure of how alleles at the two loci are associated and is given by: |
|
When r2 is = 1, there are only two possible haplotypes, and knowing the allele at locus A is completely predictive of the allele present at locus B. In this case, D′ also = 1. However, D′ can = 1 when r2 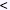 1. In this case, there will only be three possible haplotypes in the population. If D′ is < 1, there will be four haplotypes in the population. This is illustrated in the colored two-locus phased haplotype displays shown below. 1. In this case, there will only be three possible haplotypes in the population. If D′ is < 1, there will be four haplotypes in the population. This is illustrated in the colored two-locus phased haplotype displays shown below. |
 Only gold members can continue reading. Log In or Register to continue
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|