97 Familial Autoinflammatory Syndromes
The familial autoinflammatory syndromes, often referred to as hereditary periodic fever syndromes, comprise rare hereditary disorders with a common phenotype of lifelong, recurrent inflammatory episodes, characterized by inflammatory symptoms such as fever, abdominal pain, diarrhea, rash, or arthralgia.1 Between the fever episodes, patients with most of these syndromes generally feel healthy and function normally. Routine laboratory investigations during a fever attack invariably reveal a severe acute-phase response with a high erythrocyte sedimentation rate, leukocytosis, and high concentrations of acute-phase proteins such as C-reactive protein (CRP) and serum amyloid A (SAA). The inflammatory episodes occur without an obvious trigger, although some patients note a relationship to physical stimuli (e.g., exposure to cold), emotional stress, or the menstrual cycle. The episodes resolve spontaneously in days or weeks. Patients go undiagnosed for years, generating a high level of discouragement and frustration for patients and physicians when no diagnosis is made.2,3 The term autoinflammatory, coined by McDermott and colleagues in 1999,4 describes the phenotype of recurrent, acute inflammatory responses. It is preferable to the term autoimmune in these cases because typical autoimmune phenomena are not found; the defect is located more in the innate immune system than the acquired immune system.5
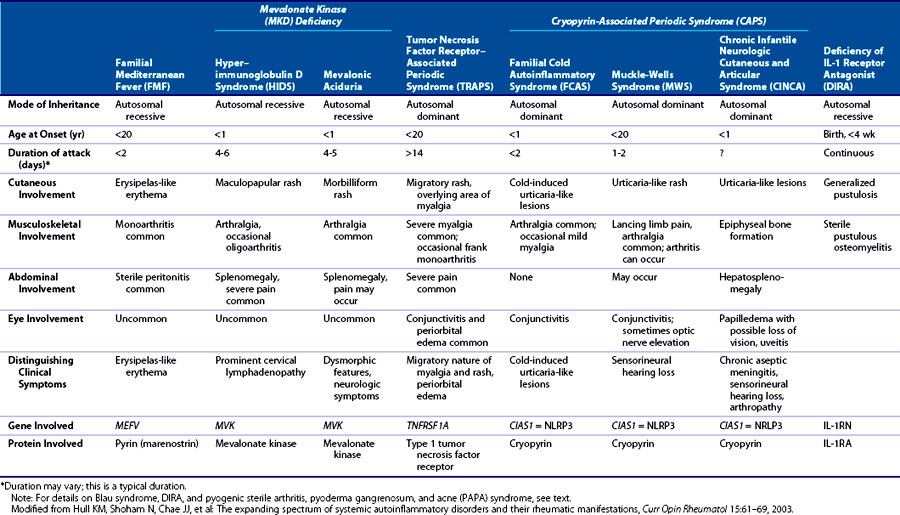
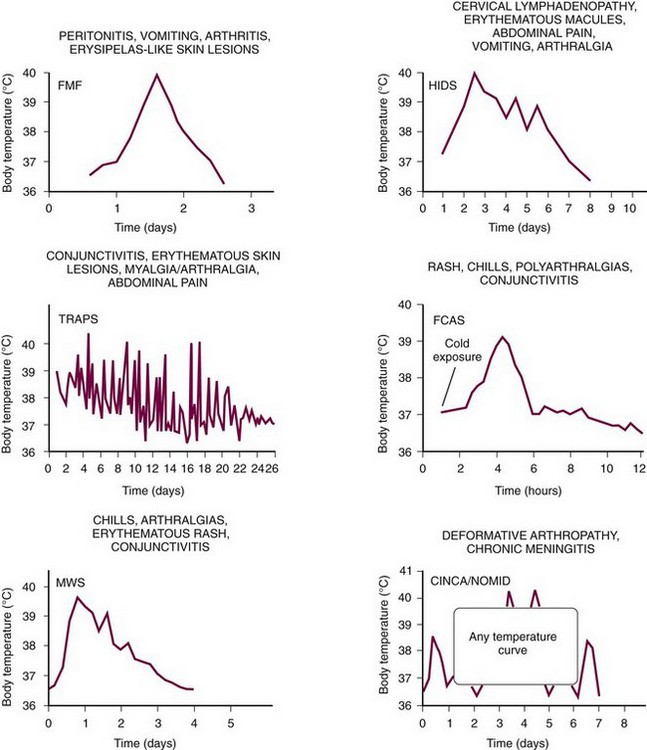
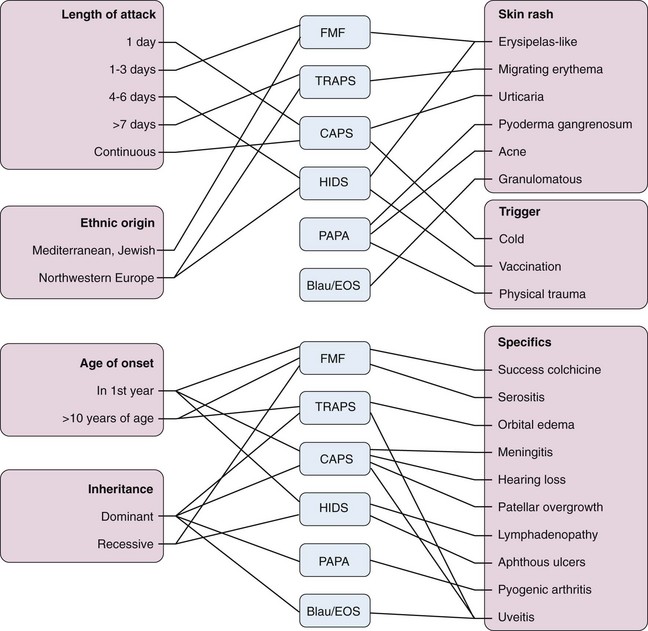
Several genetically distinct types of hereditary autoinflammatory syndromes are recognized. Despite the common phenotype described previously, these can often be differentiated clinically by specific characteristics, in particular mode of inheritance, age of onset, average duration of the fever episodes and the fever-free interval, geographic region of origin of the patient, and occurrence of long-term complications such as amyloidosis or deafness (Table 97-1 and Figure 97-1). A significant number of patients with a periodic fever phenotype still do not fit into this genetically based classification, probably representing additional (genetic) defects that can lead to autoinflammatory disease. This chapter describes the seven best characterized familial autoinflammatory syndromes at this time in detail.
Differential Diagnosis
When a patient has had recurrent fever episodes for more than 2 years, it is increasingly unlikely that these are caused by an infection or a malignant disorder. The differential diagnosis at that time may include numerous inflammatory disorders such as juvenile rheumatoid arthritis, adult-onset Still’s disease, inflammatory bowel disease, Schnitzler syndrome, and Behçet’s disease, in addition to the hereditary periodic fever syndromes (Table 97-2). Because the hereditary syndromes are rare (except for familial Mediterranean fever [FMF] in individuals with a distinct ethnic background), the more common diagnoses should usually be excluded first.
Table 97-2 Differential Diagnosis of Periodic Fever
The mainstay of the diagnosis of hereditary autoinflammatory syndromes is clinical assessment, with a detailed medical and family history, and preferably at least one observation of the patient during a fever episode because physical examination of the patient in a period of remission is seldom abnormal. Another helpful clue, although not pathognomonic, is often gained from knowing the patient’s ethnic origin. This clinical assessment often yields enough information to build a differential diagnosis of the specific familial autoinflammatory syndromes (see Table 97-1) to determine the direction of genetic testing (Figure 97-2).
Familial Mediterranean Fever
Epidemiology
FMF (Mendelian inheritance in men [MIM] 249100) is the most prevalent disorder among the hereditary autoinflammatory syndromes, with more than 10,000 patients affected worldwide. It occurs primarily in people originating from the Mediterranean basin including Armenians, Sephardic Jews, Arabs, and Turks. FMF is an autosomal recessively inherited disorder. Most families reported with an apparent autosomal dominant inheritance pattern of FMF6 represent examples of pseudodominant inheritance owing to consanguinity combined with the high carrier frequency of FMF mutations in certain populations6–8; however, at least three families studied do seem to show a true dominant inheritance, even after extensive genetic analysis.8
Etiology
In 1997 two groups independently traced the genetic background of FMF to a hitherto unknown gene on the short arm of chromosome 16, dubbed the MEditteranean FeVer (MEFV) gene.9,10 At least 80 disease-linked mutations in the MEFV gene have been described so far, most of which are clustered in the tenth exon of this gene (for details see the online mutation database at http://fmf.igh.cnrs.fr/infevers/). Most are missense mutations that produce a single amino acid change in the protein (Figure 97-3). There are six common mutations, accounting for almost 99% of all FMF chromosomes: M694V (occurring in 20% to 65% of cases, depending on the population examined11), V726A (in 7% to 35%), M680I, M694I, V694I, and E148Q. For the first three mutations mentioned here, a founder effect has been established,10 pointing to common ancestors at least 2500 years ago. The high frequency of the mutated MEFV gene in more than one Middle Eastern population has led to the hypothesis that heterozygous carriers have an as-yet-unknown advantage, possibly a heightened (inflammatory) resistance to an as-yet-unidentified endemic pathogen of the Mediterranean basin.10 In about 30% of patients, only one or no mutations in the MEFV gene can be detected; the etiology in these patients still needs to be determined.
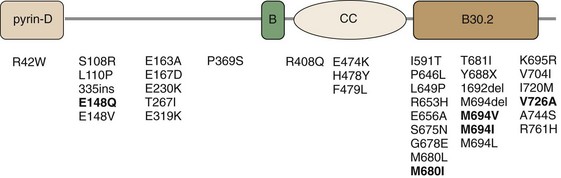
Figure 97-3 Schematic representation of pyrin (marenostrin) protein, with four conserved domains including a pyrin domain, a B-box (B), coiled-coil domain (CC), and a B30.2 domain. Indicated are mutations as found in familial Mediterranean fever, with the five most common missense mutations in bold type. For complete listing of all currently known mutations, see INFEVERS website: http://fmf.igh.cnrs.fr/infevers/.
Pathogenesis
The MEFV gene encodes for a protein of 781 amino acids, known as pyrin or marenostrin. Pyrin is expressed as a cytoplasmic protein in mature monocytes in association with microtubules12 but is predominantly found in the nucleus in granulocytes, dendritic cells, and synovial fibroblasts.13 The expression of pyrin is induced by inflammatory mediators such as interferon-α and tumor necrosis factor (TNF).14 The pyrin domain is shared by many proteins involved in apoptosis and inflammation and is a member of the death-domain superfamily that includes death domains, death-effector domains, and caspase-recruitment domains. Pyrin binds specifically to other proteins that contain a pyrin domain, which include the adapter protein “apoptosis-associated specklike-like protein with a CARD” (ASC).
The proinflammatory cytokine interleukin (IL)-1β is central in the pathogenesis of FMF. This cytokine is expressed as an inactive precursor, which is cleaved by caspase-1 to yield the active IL-1β. Caspase-1 itself first needs to be activated through the interaction with a protein complex termed an inflammasome. Several inflammasomes have been described so far. The major inflammasome complex involved in the activation of caspase-1 and IL-1β is the cryopyrin or NLRP3 inflammasome.15,16 Two hypotheses have been proposed regarding the effect of pyrin on IL-1β processing. The “sequestration hypothesis” holds that pyrin has an inhibitory effect on caspase-1–mediated activation of IL-1β, through its prevention of the formation of the cryopyrin inflammasome by competitive binding of the adapter protein ASC and procaspase-1 and binding caspase-1.17,18 Under this hypothesis, FMF mutations are thought to interfere with the inhibiting interactions of pyrin, resulting in decreased regulation of IL-1β activation.18 The second hypothesis, proposed by Yu and colleagues,19 suggests that pyrin can form its own specific inflammasome for activation of IL-1β, although not all the components of this proposed inflammasome have been specified so far. The FMF mutations would increase the sensitivity of this putative pyrin inflammasome. Apart from its role in regulation of IL-1β, there is also conflicting evidence for the effect of pyrin on regulation of nuclear factor κB (NFκB) or apoptosis, varying from inhibition to stimulation.1
Clinical Features
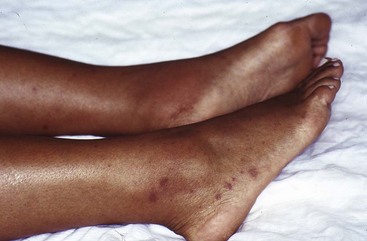
In approximately 90% of FMF patients, symptoms start before age 20 years.20 The inflammatory attacks of FMF usually last 1 to 3 days The frequency can vary widely; 2 to 4 weeks is the most common interval (see Figure 97-1). Symptoms of serositis (i.e., peritonitis, pleuritis, synovitis) are the main feature of FMF attacks, usually accompanied by fever. Abdominal pain of 1 or 2 days’ duration occurs in 95% of patients, varying in severity from severe peritonitis resembling an acute abdomen to only mild abdominal pain without overt peritonitis.21 Arthritis (rarely destructive) is often confined to one large joint such as the knee, ankle, or wrist and may be the only symptom. Chest pain resulting from pleuritis is usually unilateral and associated with a friction rub or transient pleural effusion. Skin involvement occurs in approximately 30% of patients, most often as erysipelas-like skin lesions on the shins or feet (Figure 97-4).22 Other, more uncommon, symptoms are pericarditis, occurring in less than 1%23; acute scrotal swelling and tenderness24; aseptic meningitis; and severe protracted myalgia, especially of the legs.
Diagnosis and Diagnostic Tests
FMF is still primarily a clinical diagnosis. There is a set of validated diagnostic criteria with a reported sensitivity and specificity of 96% to 99% (Table 97-3).25 These criteria were validated in a population with a high prevalence of FMF and low prevalence of the other autoinflammatory disorders, however, and the ethnic origin of the patient needs to be taken into account. In molecular diagnostic testing, genetic laboratories usually screen for the five most common mutations and rare mutations are missed. MEFV mutations occur on both alleles in only 70% of typical cases,26 whereas in the remaining 30%, only one or no mutation can be detected, even after sequencing. There is also evidence of reduced penetrance. Despite these limitations, molecular testing can be used as a confirmatory test. Whether or not the results are positive, treatment with colchicine is warranted in symptomatic cases of fitting ethnic origin fulfilling the diagnostic criteria.27,28 No specific biologic marker is available to distinguish an inflammatory FMF attack from an infectious fever or appendicitis. During an inflammatory attack, there is an acute-phase response, which includes elevation of SAA, CRP, and plasma fibrinogen and polymorphonuclear leukocytosis. Proteinuria in patients with FMF is highly suggestive of renal amyloidosis.
Table 97-3 Diagnostic Criteria for Familial Mediterranean Fever*
| Major Criteria |
| Minor Criteria |
* Requirements for diagnosis of familial Mediterranean fever are ≥1 major criteria or ≥2 minor criteria.
† Typical attacks are defined as recurrent (≥3 of the same type), febrile (≥38° C), and short (lasting between 12 hours and 3 days).
‡ Incomplete attacks are defined as painful and recurrent attacks not fulfilling the criteria for a typical attack.
From Livneh A, Langevitz P, Zemer D, et al: Criteria for the diagnosis of familial Mediterranean fever, Arthritis Rheum 40:1879–1885, 1997.
Treatment
Colchicine is the first-line treatment for patients with FMF. Colchicine prevents inflammatory attacks completely in 60% to 75% of patients, and it significantly reduces the number of attacks in an additional 20% to 30%.29 The average dose in adults is 1 mg daily, but this may be increased to 3 mg in cases in which no response is seen at the lower dose. This regimen is usually well tolerated; gastrointestinal side effects including diarrhea and abdominal pain generally resolve with dose reduction. More serious side effects such as myopathy, neuropathy, and leukopenia are rare and occur primarily in patients with renal or liver impairment. During a fever attack, oral or intramuscular nonsteroidal anti-inflammatory drugs (NSAIDs) can be used for pain relief. Glucocorticoids have limited efficacy.
Compliance with colchicine use is important because colchicine has been shown to prevent the occurrence of amyloidosis. Since the introduction of colchicine therapy, the incidence of amyloidosis in FMF has decreased dramatically, whereas in areas with a high prevalence of FMF where colchicine is not routinely available, such as Armenia, amyloidosis is still common. Colchicine’s principal effect at the cellular level is to depolymerize microtubules by interacting with tubulin, inhibiting motility and exostosis of intracellular granules. It has a powerful antimitotic effect, causing metaphase arrest. It has been speculated, in cases of infertility in patients treated with colchicine, that this medication causes azoospermia. Colchicine does not have a significant adverse effect on sperm production or function, however.30 Unfounded fear of teratogenic effects of colchicine often wrongly leads to cessation of this drug in young women who want to get pregnant, with a subsequent increased frequency and severity of attacks, which enhances problems with fertility and pregnancy. Colchicine has proved to be safe, even in early pregnancy, and treatment should not be interrupted for this reason.31,32 It can also be used while breastfeeding.29
In about 5% to 10% of patients, FMF is refractory to colchicine use. Lidar and colleagues33 used parenteral colchicine in such refractory cases, but this can be toxic. More recently, the IL-1β inhibitor anakinra has been shown to be effective in several case reports and series.34–38
Outcome
Recurrent attacks of peritonitis may lead to intra-abdominal or pelvic adhesions, resulting in complications such as small bowel obstruction. Another serious long-term complication of FMF is amyloid A (AA) amyloidosis. This amyloidosis is primarily found in the kidneys, resulting in renal failure, but can also occur in the gastrointestinal tract, liver, and spleen, and eventually in the heart, testes, and thyroid. The prevalence of amyloidosis varies, especially depending on the ethnic origin, but it is high in untreated patients. It is common among Sephardic Jews but rare in Ashkenazi Jews.39
For a variety of reasons including peritoneal adhesions and ovulatory dysfunction, subfertility in women is common.31 In men, subfertility secondary to azoospermia (sometimes secondary to testicular amyloidosis) or impairment of sperm penetration has been found.20
Hyper–immunoglobulin D Syndrome (Mevalonate Kinase Deficiency)
Epidemiology
Hyper-IgD syndrome (HIDS) (MIM 260920), also known as mevalonate kinase deficiency, is an autosomal recessively inherited disorder, but it is far less prevalent than FMF. The International Hyper-IgD Syndrome Registry, based in Nijmegen, the Netherlands, in which clinical information is collected from physicians worldwide, currently holds data on approximately 220 patients. Approximately 75% of these patients are from Western Europe, and 50% are from the Netherlands and France.40 Most HIDS patients are of Caucasian origin. These observations can be explained partly by a founder effect.41 In the Netherlands, the carrier frequency of the most common mevalonate kinase mutation is 1 : 153.42 Men and women are affected in equal numbers.40
Etiology
HIDS is caused by mutations in the gene encoding for the enzyme mevalonate kinase, located on the long arm of chromosome 12 (for details, see the online mutation database available at http://fmf.igh.cnrs.fr/infevers/).43–45 Patients with classic HIDS are most often compound heterozygotes for two missense mutations (Figure 97-5). Two mutations (V377I and I268T) account for more than 85% of the patients described to date.40
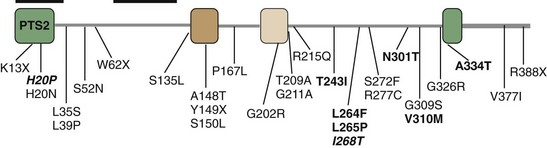
Figure 97-5 Mevalonate kinase, with four conserved domains represented by colored boxes. Indicated are missense mutations, nonsense mutations, and two deletions, which have been identified in mevalonate kinase deficiency. In bold are mutations found in mevalonic aciduria patients; in bold and italic are mutations found in classic hyper-IgD syndrome and mevalonic aciduria. For complete listing of all currently known mutations, see INFEVERS website: http://fmf.igh.cnrs.fr/infevers/.
The term variant type HIDS, which we proposed for patients with an autoinflammatory disease and high immunoglobulin (Ig)D without mevalonate kinase deficiency,45 has been largely abandoned. Many patients with fever syndromes have a raised IgD and this subclass is heterogeneous. It seems more useful to designate these patients as “autoinflammatory disease not otherwise specified” to indicate that a more specific diagnosis may still be found in the future.
Pathogenesis
Mevalonate kinase is part of the isoprenoid pathway; it is the step after 3-hydroxy-3-methyl-glutaryl–coenzyme A (HMG-CoA) reductase, phosphorylating mevalonic acid. The isoprenoid pathway has many diverse end products that include cholesterol, dolichol, and ubiquinone, and it leads to isoprenylation of proteins, with a post-translational modification directing these proteins such as Rho and Ras to the cell membrane.46
HIDS mutations lead to a constantly diminished activity of mevalonate kinase to about 5% to 15% of normal levels, and these levels decrease further during a fever attack.47 Because of this reduced enzyme activity, the substrate mevalonic acid accumulates in serum and urine. Higher levels are found during the episodes of fever. There does not seem to be a dramatic shortage of any specific end product; concentrations of cholesterol, ubiquinone, and dolichol in patients are normal to slightly decreased.1
Another syndrome was already linked to mutations in the mevalonate kinase gene before the discovery of HIDS48—classic mevalonic aciduria. Patients with mevalonic aciduria carry specific mutations that cause a more severe reduction of mevalonate kinase enzyme activity, reducing it to undetectable levels. These patients constantly produce large amounts of mevalonic acid and often have more than 1000 times as much mevalonic acid in their urine than do HIDS patients.49 Patients with mevalonic aciduria also have a more severe phenotype. Classic mevalonic aciduria and HIDS seem to be two extremes of a continuous spectrum of disease related to mevalonate kinase deficiency.50
The pathogenetic link between mevalonate kinase deficiency and inflammation is still unclear, but there is increasing evidence for a connection between the isoprenoid pathway and inflammation. Inhibition of the isoprenoid pathway by statins, the inhibitors of HMG-CoA reductase (the enzymatic step before that of mevalonate kinase), can have anti-inflammatory effects, ranging from increased apoptosis of inflammatory cells to reduction of expression of cytokines.51,52 In other settings, statins seem to be proinflammatory, most notably in a study in which stimulation with Mycobacterium tuberculosis or mitogens in combination with statins increased caspase-1 activation and IL-1β secretion by monocytes, through a decrease of geraniolgeraniol.15 The ex vivo production of IL-1β is increased in HIDS,53,54 whereas treatment with the IL-1 blocker anakinra is beneficial.55,56 Current evidence points to a link between the mevalonate pathway and IL-1 through alterations of isoprenylation of the small GTPase Rac1, phosphoinositide 3-kinase (PI3K), and protein kinase B (PKB).57 A defect in apoptosis may also contribute to the pathogenesis of HIDS. Lymphocytes from HIDS patients (who had no fever at the time of blood sampling) showed a decrease in apoptosis when stimulated with anisomycin, which was not found in patients with TNF receptor–associated periodic syndrome (TRAPS) or FMF patients.58 Such a decrease in apoptosis would result in increased survival of lymphocytes and may delay the resolution of the inflammatory response. An ordinarily innocuous stimulus in HIDS patients would more easily lead to a full-blown fever episode.
Clinical Features
Ninety percent of patients with HIDS experience their first fever episode in the first year of life,40 and these episodes become most frequent in childhood and adolescence. The high fevers may lead to seizures, especially in young children. Vaccination, minor trauma, surgery, and physical or emotional stress are factors that provoke a fever episode, although often a triggering factor is not obvious.40 The fevers often begin with cold chills and a sharp increase in body temperature. They are almost always accompanied by cervical lymphadenopathy and abdominal pain with vomiting and diarrhea. Other frequent symptoms are headache, myalgia, and arthralgia. Apart from the lymphadenopathy, physical signs frequently consist of splenomegaly and a skin rash with erythematous macules and papules (Figure 97-6) or petechiae (Figure 97-7).40 Sometimes there are also signs of frank arthritis (principally large joints) and hepatomegaly. About 40% of patients report painful aphthous ulcers in the mouth, vagina, or scrotum (Figure 97-8). The fever disappears spontaneously after 3 to 5 days, although it may take longer before the symptoms in joints or skin disappear completely. These inflammatory attacks occur, on average, once every 4 to 6 weeks, although this may vary from patient to patient or in an individual patient.
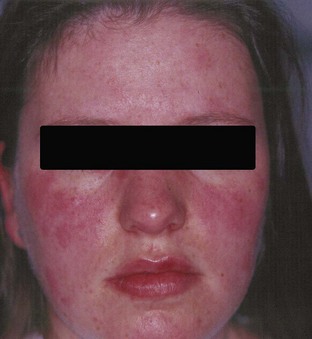
Figure 97-6 Facial erythematous macules and papules in a hyper-IgD syndrome patient during an attack.
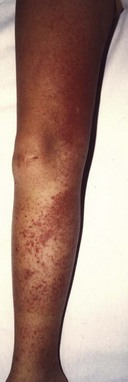
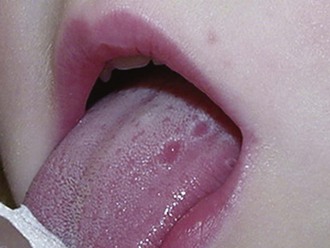
Figure 97-8 Aphthous ulceration detected on the tongue of a patient with hyper-IgD syndrome.
(Courtesy Dr. K. Antila, North Carelian Central Hospital, Joensuu, Finland.)
Patients with mevalonic aciduria, the metabolic disorder that is also caused by mevalonate kinase gene mutations, experience similar but more severe inflammatory episodes as HIDS patients. In addition, these patients suffer from psychomotor retardation, ataxia, failure to thrive, cataracts, and dysmorphic facies. Patients with classic mevalonic aciduria usually die in early childhood.49 An intermediary clinical phenotype between classic mevalonic aciduria and HIDS has been described.50
Diagnosis and Diagnostic Tests
HIDS is diagnosed on the basis of a combination of characteristic clinical findings and continuously elevated IgD concentrations (>100 IU/mL) (Table 97-4). There are numerous caveats concerning IgD serum concentration, however: Values may be normal in young patients (especially patients younger than 3 years old),59 persistently normal levels have been reported in a few patients with HIDS,43 and patients with other familial autoinflammatory syndromes may also have elevated IgD concentrations, although these are usually only slightly elevated. More than 80% of HIDS patients combine a high concentration of IgD with high IgA levels.59,60 During fever attacks, a brisk acute-phase response is observed including leukocytosis, high levels of SAA and CRP, and activation of the cytokine network.53,61
Table 97-4 Diagnostic Indicators of Hyper–Immunoglobulin D Syndrome
| At Time of Attacks |
| Constantly Present |
| Specific Features |
* Extremely high serum concentrations of IgD are characteristic but not obligatory.
The diagnosis of HIDS can be confirmed by DNA analysis of the mevalonate kinase gene. The best approach is to start with screening for the two most prevalent mutations, V377I and I268T. If this screening is negative, but the clinical suspicion remains high, sequencing of the entire gene can be considered. A good alternative is the measurement of urinary mevalonic acid concentrations during an attack, which are slightly elevated. Gas chromatography–mass spectroscopy is necessary to detect this slight increase, however.62 The measurement of mevalonate kinase enzyme activity is complicated and time-consuming and should be reserved for research purposes.
Treatment
There is no established treatment regimen for HIDS. Anakinra is effective in reducing disease severity in a number of case reports55,56,63–65 and is currently the most promising therapy. A double-blind, placebo-controlled, crossover trial of the HMG-CoA reductase inhibitor simvastatin showed a beneficial effect of this drug, with a reduction in number of days of illness in five out of six patients66; however, in clinical practice the beneficial effect is not impressive. Favorable preliminary experience with the TNF antagonist etanercept has been reported.55,67,68
Some individual patients have been reported to have benefited from treatment with corticosteroids, colchicine, intravenous immunoglobulin, or cyclosporine, but these results have not been repeated in most patients.40 Thalidomide did not have an effect on disease activity in a placebo-controlled trial.69
Outcome
The long-term outcome in HIDS is relatively benign in most patients. In some patients, the fever episodes occur less frequently and become less severe later in life, starting from late adolescence.40 Joint destruction is rare, but abdominal adhesions are seen, resulting from repeated abdominal inflammation or (unnecessary) diagnostic laparotomy because of suspected acute abdomen.
Until more recently, no cases of amyloidosis had been seen in HIDS patients since its first description in 1984. Since 2004, four HIDS patients 19 to 27 years old have been reported who developed renal failure because of AA amyloidosis.40,70–72 Regular screening for proteinuria also may be advisable in HIDS patients, especially patients with frequent and severe fever episodes.
Tumor Necrosis Factor Receptor–associated Periodic Syndrome
Epidemiology
TRAPS (MIM 142680) has an autosomal dominant inheritance pattern. It was originally described in a large family from Irish and Scottish descent as “familial Hibernian fever.”73 It is found primarily in patients from northwestern Europe but also has been described in families from Australia, Mexico, Puerto Rico, Portugal, and the Czech Republic.74 Any ethnic group may be affected. Other abandoned nomenclature for this syndrome includes “autosomal dominant familial periodic fever”75 and “familial perireticular amyloidosis.”76
Etiology
Mutations are found in the gene for the type I TNF receptor (TNFRSF1A), which is located on the short arm of chromosome 12.4 These are mainly single-nucleotide missense substitutions, located in exons 2, 3, and 4, which encode for the extracellular domain of TNFRSF1A. Many of these mutations disrupt one of the highly conserved cysteine residues involved in extracellular disulfide bonds of the 55-kD type I TNF receptor protein (Figure 97-9) (for details, see the online mutation database available at http://fmf.igh.cnrs.fr/infevers/).77
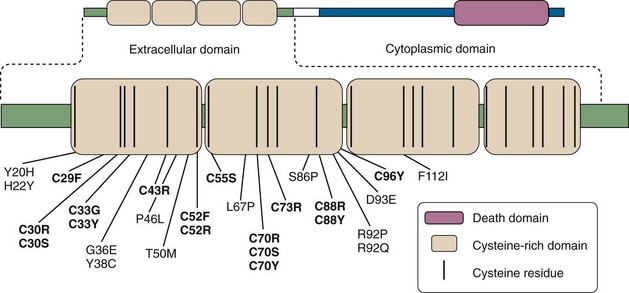
Figure 97-9 Schematic representation of the tumor necrosis factor (TNF) receptor type 1 protein (TNFRSF1A), depicting mutations found in TNF receptor–associated periodic syndrome up to this time (except for one intron mutation affecting a splice site). Mutations disrupting cysteine residues are in boldface type. For complete listing of all currently known mutations, see INFEVERS website: http://fmf.igh.cnrs.fr/infevers/.
There are some general genotype-phenotype correlations, especially when mutations are grouped in cysteine and noncysteine mutations. Noncysteine mutations have, overall, a lower penetrance than cysteine mutations, and amyloidosis is seen far more often in association with cysteine mutations.78 Two missense mutations in TNFRSF1A, P46L and R92Q, have a particularly low penetrance and are found in approximately 1% to 10% of control chromosomes.78–80 R92Q has been observed in higher prevalence in a group of patients with arthritis. It is thought that the clinical manifestations of patients with an R92Q mutation depend on other so-far-unidentified modifying genes, environmental factors, or both.77,78
Pathogenesis
TNF is a pleiotropic molecule, which induces cytokine secretion, activation of leukocytes, fever, and cachexia. Activation of the TNF receptor by TNF causes cleavage and shedding of its extracellular part into the circulation, where it acts as an inhibitor of TNF. However, TRAPS-associated mutations in the TNF receptor lead not to an increase but rather to a loss of TNF-signaling function including less binding of TNF,81,82 less cell surface expression,82–85 and decreased TNF-induced NFκB-activation.84,86,87 The mutated TNFRSF1A is retained intracellularly, pooled in the endoplasmic reticulum.82,83,87,88 Mutant TNFR1 cannot associate with the wild-type version but can form aggregates by self-interaction.82,83 This cytoplasmic receptor aggregation results in ligand-independent signaling.87,89 Mitochrondrial-derived reactive oxygen species appear to mediate this effect.90 This new hypothesis also might offer an explanation for the observation that blocking IL-1β works better in some TRAPS patients than blocking TNF.87,91
An alternative hypothesis, the “shedding hypothesis,” was postulated on the finding of reduced shedding, which leads to prolonged TNF signaling and uncontrolled inflammation. Not all TRAPS mutations cause decreased shedding, however, and although serum concentrations of the shedded soluble TNFRSF1A in TRAPS patients during periods without symptoms are often found to be significantly reduced compared with normal subjects, this is not always the case.1 The hypothesis of reduced shedding, although attractive by its simplicity, is not supported as the sole cause of the fever attacks in TRAPS, and additional mechanisms seem to be at work.
Clinical Features
The clinical features can vary much more between individual TRAPS patients than is generally seen in FMF or HIDS.74,77 The age of onset can vary, even within the same family, with a documented range of 2 weeks to 53 years old.74,92 There is also a large variation in duration and frequency of the fever episodes in TRAPS. On average, attacks last 3 to 4 weeks and recur two to six times each year, but episodes also may be limited to a few days (see Figure 97-1). Although the index patient, through whom the diagnosis is made, often displays well-defined inflammatory attacks, affected family members may have less typical symptoms such as episodic mild arthritis.
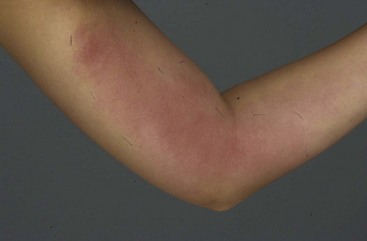
During inflammatory attacks, a high, spiking fever can be accompanied by skin lesions, myalgia and arthralgia, abdominal distress, and ocular symptoms. The most common cutaneous manifestation is a centrifugal, migratory, erythematous patch, which may overlie a local area of myalgia (Figure 97-10),93 but urticarial plaques also may be seen. Myalgia is often located primarily in the muscles of the thighs, but it may migrate during the fever episode, affecting all of the limbs and the torso, face, and neck. Arthralgia primarily affects large joints including hips, knees, and ankles. Frank synovitis is rarer, and when it does occur it is nonerosive, asymmetric, and monoarticular.74 Abdominal pain occurs in 92% of TRAPS patients during inflammatory attacks; other gastrointestinal symptoms often seen include vomiting and constipation. Ocular involvement is characteristic in TRAPS, and it may involve conjunctivitis, periorbital edema, or periorbital pain in one or both eyes. Severe uveitis and iritis have been described, and any TRAPS patient with ocular pain should be examined for these complications.74,93 Other, less frequently observed symptoms during fever attacks in TRAPS are chest pain, breathlessness, pericarditis, and testicular and scrotal pain, which may be caused by inflammation of the tunica vaginalis. One case report described a patient who presented with psychosis without fever.94 It has been suggested from observation in one of the first families with TRAPS that this disorder is associated with an increased incidence of indirect inguinal hernias,95 but this has not been shown in other patients. Lymphadenopathy is rare in TRAPS.
Diagnosis and Diagnostic Tests
As in the other familial autoinflammatory syndromes, laboratory investigations during inflammatory attacks show a clear acute-phase response, and even in between fever attacks, such an inflammatory response may be measured. The IgD level may be elevated, but the value is almost always less than 100 IU/mL.92,95
Hull and colleagues74 proposed a set of clinical diagnostic criteria for TRAPS (Table 97-5). These criteria are not validated by epidemiologic measures, but they may be used as a first step in evaluation of patients. TRAPS is ultimately a genetic diagnosis, defined by a missense mutation in the gene for TNFRSF1A. Clinical penetrance of TRAPS mutations is not 100%, however, even for cysteine mutations, and asymptomatic carriers are common. Also, the finding of an R92Q or P46L variant in this gene would pose a difficulty. Because they have many characteristics of a polymorphism rather than a direct disease-causing mutation (see etiology), it is debatable whether such a finding should lead to a diagnosis of TRAPS.
Table 97-5 Diagnostic Indicators of Tumor Necrosis Factor Receptor–Associated Periodic Syndrome
1. Recurrent episodes of inflammatory symptoms spanning >6 mo duration (several symptoms generally occur simultaneously) < div class='tao-gold-member'> Only gold members can continue reading. Log In or Register to continue
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
