22 Epigenetics
The term epigenetic describes a number of chromatin modifications that stably alter gene expression without changing the sequence of the DNA. In this way, epigenetic mechanisms shape the phenotype of a cell without changing the genotype. In the last few years, the study of epigenetic mechanisms strongly progressed, yielding insight into the mechanisms by which epigenetic chromatin modifications regulate gene expression. It became clear that epigenetic mechanisms are important in cellular development and differentiation, as well as in adaptation to changes in the environment. In particular, Conrad Waddington is given credit for advancing the hypothesis that the environment plays a sculpting role in development and cell fate via epigenetic mechanisms.1 Epigenetics supports the idea that changes in gene expression that stem from long-term exposure to a certain signal get imprinted, become independent of the activating stimulus, and persist even in its absence. Accordingly, the impact of nutrition, toxins, or infection on gene expression is maintained, leading to fixed changes in the phenotype. This change helps the cell to adapt but might also promote pathologic cellular behavior.
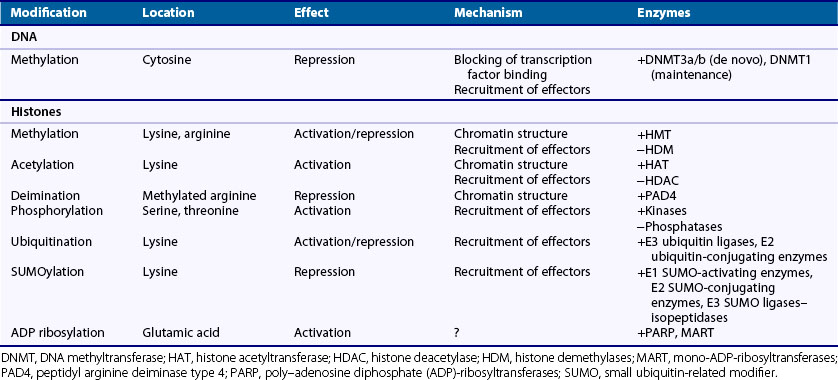
With the acquisition of new knowledge, the original definition of epigenetic processes as heritable changes in DNA without alterations in the sequence became a matter of debate. Particularly divisive is the matter of heritability. Even though the classic definition demands that epigenetic modifications persist through mitosis and meiosis (i.e., are inherited to the next generation), transient modifications that indirectly initiate stable changes more and more are put together under the term epigenetics. Giving credit to this view, Bird described epigenetic events as “the structural adaption of chromosomal regions so as to register, signal, or perpetuate altered activity states.”2 In addition to the well-established epigenetic role of DNA methylation, this definition includes a variety of more transient histone modifications such as acetylation, methylation, or phosphorylation that underlie epigenetic effects, and that will be discussed in this chapter along with the influence of SUMOylation, ubiquitination, adenosine diphosphate (ADP) ribosylation, and microRNA.
Epigenetic Mechanisms
DNA Methylation
DNA methylation is the classic and probably best studied epigenetic modification. Thereby, methyl marks are added to the cytosine ring of cytosine-phosphatidyl-guanine (CpG) dinucleotides. In addition, recently also non-CpG, but CHG or CHH, where H stands for A, C, or T, DNA methylation has been found to occur.3 This type of DNA methylation seems to be important mainly in pluripotent stem cells and disappears during differentiation. CpG methylation of DNA preferably is found at sites with a high percentage of CpGs, so-called CpG islands. Recently however, differences in the methylation pattern between tissues were found to occur frequently not at CpG islands, but at regions with lower CpG density that lie around 2 kb upstream or downstream of CpG islands, so-called CpG island shores.4 At both locations, DNA methylation leads to transcriptional repression.
During embryonic development, de novo methylation marks are established by DNA methyltransferase (DNMT) 3a and 3b. During somatic cell replication, methylation marks are stable and are maintained by DNMT1. Gene silencing by DNA methylation is crucial for the formation of heterochromatin. It promotes stable silencing of repetitive sequences such as alu sequences and retrotransposons, genomic imprinting, X chromosome inactivation in female mammals, and tissue-specific gene expression. However, in recent years it has become clear that DNA methylation is more dynamic than was previously assumed. Global genomic demethylation is seen in the paternal pronucleus shortly after fertilization, and gene-specific demethylation has been observed.5 Promoters of estrogen-responsive genes, for instance, are periodically methylated and demethylated depending on ligand binding of the estrogen receptor α.6,7 Methylation marks are believed to be removed passively (e.g., DNMT1 does not restore methyl marks after replication) or actively. Different mechanisms have been proposed to account for active DNA demethylation; however, as yet definite experimental proof in mammalian cells exists for none of them.8 In short, conceivable mechanisms for active DNA demethylation include enzymatic removal of the methyl group, excision and substitution of 5-meC by C, oxidative demethylation thereby converting 5-meC to 5-hydroxymethylcytosine, and demethylation by S-adenosylmethionine (SAM) domains, forming a 5-meC radical.
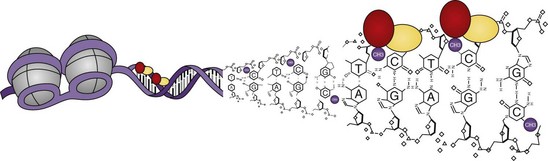
DNA methylation can directly interfere with binding sites of transcription factors, thereby inhibiting induction of gene expression, or transcriptional repressors can bind to methylated DNA, blocking transcription. By their presence, they inhibit the binding of transcription factors and recruit histone deacetylases, which change the histone code and further block gene expression (Figure 22-1).
Histone Modifications
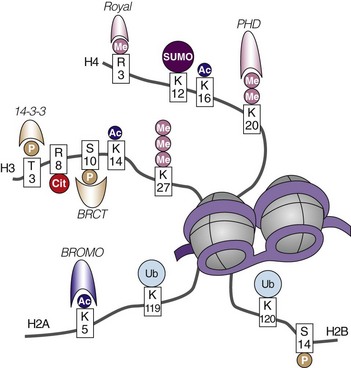
Within the nucleus, DNA is packaged around an octamer comprising homodimers of four different histone proteins: H2A, H2B, H3, and H4. This so-called nucleosome is stabilized and kept in place by H1. Each histone protein has an N-terminal tail, which is the main target of a variety of histone modifications. Amino acids in the histone tails can be acetylated, methylated, phosphorylated, ubiquitinated, SUMOylated, ADP ribosylated, and deiminated (Figure 22-2). Not all of these modifications are equally well studied, and detailed knowledge about their regulation and interactions is still missing. The sum of the different histone modifications at a distinct region of the genome is called the histone code. It is generally believed that the histone code can influence transcriptional activity directly by affecting chromatin structure, thereby making it more or less accessible for transcription factors and indirectly leading to the attraction of effector molecules that in turn recruit and stabilize the transcription machinery.9 For most histone modifications, it is yet impossible to predict whether they enhance or restrict transcriptional activity and how this will influence the biologic output. Also, more and more accumulating evidence indicates that the different histone modifications are strongly interconnected and can enforce or counteract each other. Nevertheless, some modifications can clearly be associated with transcriptionally active or inactive chromatin (Table 22-1).
Acetylation
Histone acetylation generally is associated with transcriptional activation. It is believed that histone acetylation directly opens the chromatin structure, allowing easier access to the transcription machinery.10 Acetylation loosens the interaction of the negatively charged DNA with the positively charged lysine by neutralizing the charge of lysine.11 In addition, acetylated histone tails are bound by bromodomain effectors.12 Members of the bromodomain family support protein-protein interactions to assemble a multicomponent complex involved in transcriptional activation. Histone acetylation is catalyzed by histone acetyltransferases (HATs) and is counteracted by histone deacetylases (HDACs). Up to now, two main groups of HATs—type A and type B—have been described, whereby the nuclear type A HATs are those believed to influence transcriptional activity.13 The family of HDACs comprises four classes. Classes I, II, and IV consist of 11 HDACs—HDAC1 to HDAC11—that are zinc dependent; class III includes the nicotinamide adenine dinucleotide (NAD)-dependent sirtuins, SIRT1 to SIRT7. HDACs and HATs not only modify histones, they also form complexes with transcription factors, oncoproteins, and tumor suppressors and regulate their activity by changing the acetylation status.14
Methylation
Histone methylation marks are highly complex because histones can be monomethylated, dimethylated, or trimethylated; in addition to lysine, arginine residues can be modified. Depending on the methylated site, histone methylation leads to transcriptional activation or repression. A prominent example of this divergent effect is trimethylation of histone 3 at the fourth lysine (H3K4me3), which is associated with active genes, in contrast to H3K27me3, which is found at silenced genes.15,16 It is interesting to note that the location of the methyl mark in relation to the regulated gene plays a role. Thus H3K9 methylation at the promoter region silences transcription, but within the coding region, H3K9 methylation was found to be associated with actively transcribed genes.17 Similar to histone acetylation, methylation of histone tails changes chromatin configuration and binds effector proteins. Methyl marks are recognized by plant homeodomain (PHD) fingers and the Royal-superfamily of chromatin-binding proteins, including chromodomain, tudor, and MBT domains.9 Histone methylation is catalyzed by histone methyl transferases (HMTs). The various members of this group of enzymes are highly specialized in methylating selected lysines or arginines to a certain degree (monomethylation, dimethylation, trimethylation).13 Lysine demethylases that counteract the action of HMTs on methylated lysines may contain an amine oxidase or a Jumonji C (JmjC) domain. Up to now, no arginine demethylase has been found that can reverse methylated arginine to arginine. Peptidylarginine deiminase 4 (PAD4) converts methylated arginine to citrulline, and this leads to transcriptional repression.18 Because in this step also the imine group of the arginine is removed, this reaction is termed deimination.
Phosphorylation
Histone phosphorylation is a fast way to induce gene expression, particularly after activation of mitogen-activated protein kinase (MAPK) pathways.19 Even though histone phosphorylation itself seems to be rather transient, reports suggest that histone phosphorylation influences other histone modifications such as acetylation and methylation, thereby indirectly inducing prolonged changes in the histone code.20,21 Phosphorylation of histones occurs at serine and threonine residues and is mediated by kinases and removed by phosphatases.22 Similar to acetylation, phosphorylation opens the chromatin structure by adding a negative charge to the histone but also leads to the attraction of effector proteins. The best studied effector proteins binding phosphorylated histones are 14-3-3 proteins and BRCA1 C-terminus (BRCT) domains.9
SUMOylation
E1 SUMO-activating enzymes, E2 SUMO-conjugating enzymes, and E3 SUMO ligases are the enzymes that add small ubiquitin-related modifiers (SUMOs) to histone lysines.23 SUMOylation can be reversed by isopeptidases. Even though details about gene regulation after histone SUMOylation are still unclear, it is known that SUMOylation mostly represses gene expression in close cooperation with other histone modifications.24 As a counterregulatory mechanism, SUMO marks are increased after histone acetylation and recruit HDACs to remove acetylation.24 Furthermore, SUMOylated histones associate with heterochromatin protein (HP)-1, a methyl-lysine binding protein that interacts with HMTs to repress transcription.25
Ubiquitination
Ubiquitin ligases and proteases control histone ubiquitination and deubiquitination, respectively. Ubiquitination of histone 2A and 2B generally activates gene transcription. However, other reports show that histone ubiquitination can repress transcriptional activity.26 Similar to SUMOylation, ubiquitination is strongly connected to other histone modifications and mainly alters gene expression via recruitment of effector molecules, which subsequently results in histone methylation.27
ADP Ribosylation
Histones can be mono-ADP ribosylated by mono-ADP-ribosyltransferases (MARTs) or poly-ADP ribosylated by poly-ADP-ribosyltransferases (PARPs).28 Little is known about this histone modification, and neither site specificity nor function has been clearly defined. However, synergy between histone acetylation and ADP-ribosylation has been described.29
Epigenetics and Noncoding RNAs
Noncoding RNAs (ncRNAs) are transcribed RNA molecules that do not translate into protein. Long known ncRNAs include transfer RNA (tRNA) and ribosomal RNA (rRNA). More interesting with regard to epigenetic modifications, however, are the more recently discovered long ncRNAs such as the Xist (X inactive specific transcript) transcript and small ncRNAs such as microRNAs (miRs). In female cells, Xist is transcribed from the later inactivated X chromosome (Xi). It specifically binds to Xi and promotes its silencing by histone ubiquitination, methylation, and loss of acetylation.30 Recently, another long ncRNA named Air has been shown to interact with a specific set of genes and to recruit the chromatin-modifying machinery to these loci.31 In plants and yeast, multiple examples of interaction between ncRNAs and epigenetic chromatin modifications have been found.32–34 Overall these data suggest that recognition of complementary DNA sequences by ncRNAs might guide epigenetic modifications to specific genomic loci.
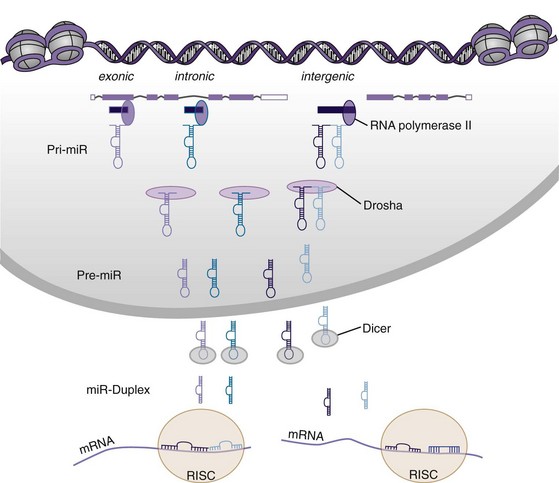
MicroRNAs are small ncRNAs that are encoded in intergenic regions and overlapping introns or exons of other genes.35 The primary miR transcript (pri-miR) containing the information of a single miR or an miR cluster is further processed by an enzymatic complex that includes the RNase III enzyme Drosha. After formation of a hairpin, the precursor miR (pre-miR) is exported to the cytoplasm (Figure 22-3). Pre-miRs are further cleaved by the RNase III enzyme Dicer and are released as short duplexes, on average 22 oligonucleotides long, which constitute the mature miR. From this duplex, two mature miRs can be formed. However, dependent on the thermodynamic stability of each end, in some cases one of the strands is fast degraded; this is usually called the miR star (*) or passenger strand.36 The other strand, termed the guide strand, is more stable and biologically active. In other cases, both strands are equally stable, and the miR from the 5′ end is referred to as 5p, whereas the miR from the 3′ end is denominated 3p. After incorporation into the RISC (RNA-induced silencing complex), mature miRs bind to the 3′ untranslated region of their target mRNA and modulate their expression by degradation or by inhibition of protein translation. The system is highly promiscuous, so that one miR can modulate expression of multiple proteins, and expression of one protein is influenced by a variety of different miRs. Mainly the miR sequence from base 2 to base 8 or 9 from the 5′ end, the so-called seed region, plays a crucial role in target recognition.37 In addition, the position of the complementary sequence on the gene and the presence of other miR target sites influence the repressive action of miRs.38 The expression of around 30% of all genes is believed to be regulated via miRs.39
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


