Electrodiagnostic Evaluation of the Peripheral Nervous System
Lawrence R. Robinson
INTRODUCTION
The ability to perform high quality electrodiagnostic studies is a critical skill for the practicing physiatrist. Electrodiagnosis is an important and helpful extension of the physical examination and can detect minor abnormalities when physical examination cannot. Moreover, the electrodiagnostic examination has several other key functions in the assessment of peripheral nervous system injuries. These techniques provide insight into the pathophysiology of a nerve lesion (such as demyelination or axon loss). By quantifying the degree of axon loss, one can estimate prognosis for recovery of lower motor neuron lesions. Finally, electrodiagnostic methods enable one to quantitatively track disease processes and complement the qualitative estimates afforded by the clinical examination.
PREPARING FOR THE ELECTRODIAGNOSTIC EXAMINATION
The electrodiagnostic examination needs to be interpreted in the context of the clinical presentation. Simply performing nerve conduction studies or needle electromyography (EMG) without understanding the patient’s presentation or developing a differential diagnosis does a disservice to the patient, as the diagnosis will often be incomplete or incorrect.
For these reasons, there are several key steps to prepare for the electrodiagnostic examination, including review of referral materials, eliciting the patient’s history, performing a directed physical examination, developing a differential diagnosis, and putting together a plan for the electrodiagnostic evaluation.
Review of referral materials: It is critical that the electrodiagnostic consultant starts by reviewing the question from the referring physician so that he or she knows what is being asked for. Without reviewing this information first, it is easy to be mislead into another diagnostic pathway and never answer the referring physician’s question. If there is any uncertainty about what is being asked for, it is often helpful to talk to the referring physician. If it is not possible to address the question being asked by the electrodiagnostic examination, one should have that discussion with the referring physician before starting the electrodiagnostic study.
Patient history: A focused patient history is very important to allow one to establish a differential diagnosis before heading into the physical examination and then the electrodiagnostic evaluation. The history should start off with the chief complaint, but should also include exacerbating or relieving factors and past medical history. It is often useful to ask what medications the patient is taking so that one is not later surprised by easy bleeding (in the setting of warfarin usage) or by a polyneuropathy (e.g., diabetes). Parts of the past medical history may be especially contributory to the differential diagnosis such as diabetes, history of cancer, or toxic exposures. When one starts to consider diseases that may be hereditary, it is always wise to ask about any family history of neuromuscular problems.
The physical examination needs to be focused and adapted to the individual presentation of the patient. Typically, it is useful to assess muscle strength not only in the involved body part but also in bilateral upper and lower limbs. One will want to detect even subtle weakness in the patient before starting the study. Since most patients coming to the EMG laboratory will have at least 4/5 strength, it is usually not possible to fully test the ankle plantarflexors or dorsiflexors by manual muscle testing. It is much more effective to have the patient walk on his or her heals and toes and do other functional tests as appropriate (e.g., squatting).
One should get a rough sense of the distribution of sensory loss. Two techniques are particularly helpful in this regard. First, in mild sensory loss, it is often helpful to compare with an area of normal sensation and ask, “If this is 100 (testing normal area), how much is this (testing abnormal area)?” Generally, losses of small percentages are not important. Secondly, if an area of abnormal or absent sensation is clearly established, it is useful to map this out, putting small pen marks where sensation changes from abnormal to normal. When discussing the distribution of sensory loss, it is better to describe it in anatomical terms (e.g., small finger) rather than in root or nerve distribution terms, since one often does not know whether sensory losses are in a peripheral nerve, plexus, root, or other distribution (e.g., ulnar nerve, medial cord, lower trunk, C8 root or spinal cord).
Muscle stretch reflexes are one of the few objective indicators of nerve or muscle disease. Hyperreflexia usually suggests an upper motor neuron etiology, while diminished reflexes or areflexia are more consistent with a peripheral nerve or lower motor neuron process.
Provocative signs can be somewhat helpful but have limited diagnostic accuracy. One may consider Spurling’s sign for cervical radiculopathy, straight leg raise sign for lumbar radiculopathy, Tinel’s, Phalen’s, or flick sign for carpal tunnel syndrome, and other maneuvers. Generally, these provocative signs have a limited specificity (1).
In many settings, where an electronic medical record is present, it is possible to briefly review radiologic findings and other laboratory data before establishing the differential diagnosis. This can be helpful to either expand or appropriately contract the differential diagnosis as needed. One should keep in mind, however, that there will not always be a good correlation between imaging studies and electrodiagnostic findings.
After completing the clinical assessment above (which should usually take no more than 15 to 20 minutes), one should establish the differential diagnosis. This important step is often skipped and can result in overlooking important possible diagnoses or missing significant tests. The examining physician should ideally write down his or her differential diagnosis. One can consider the differential diagnosis starting from most likely to least likely or starting centrally in the nervous system and work peripherally so that important possibilities are not overlooked. It is generally wise to include the diagnosis the referring physician has suggested on your list, since you want to be able to answer his or her question in your final report.
After the differential diagnoses have been articulated, one can then establish a plan for diagnostic testing. One should look at the differential diagnoses and decide what elements of electrodiagnostic study are needed to assess for those possible diagnoses. One may elect to modify this plan as initial information is attained, and in fact, this is often an iterative process. However, without going through the rigor of developing (and writing down) a plan based on the differential diagnosis, one will miss important findings (Fig. 4-1).
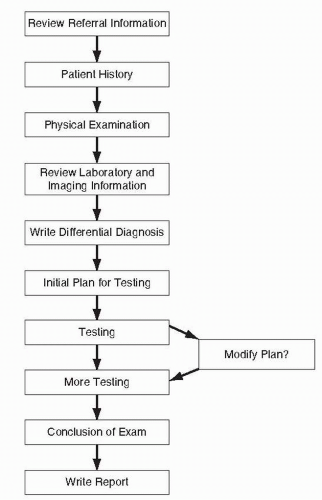 FIGURE 4-1. Steps in the evaluation of a patient referred for an electrodiagnostic medicine consultation. |
TYPES OF ELECTRODIAGNOSTIC TESTING
Although there are many techniques that can be employed in electrodiagnostic assessment, this chapter will cover the primary techniques including nerve action potentials (sensory and mixed), motor nerve conduction studies, late responses (F waves, H waves, and A waves), needle EMG, and repetitive stimulation. For each technique, this chapter will discuss the physiology, normal findings, applications, abnormalities, limitations, and the likely encountered sources of error.
Sensory Nerve Action Potentials and Compound Nerve Action Potentials
Sensory nerve action potentials (SNAPs) and compound nerve action potentials (CNAPs) both involve electrical stimulation of nerves and recording of the synchronized discharge of axons within the nerve at some distance from the point of stimulation. SNAPs are responses that only involve sensory axons, whereas CNAPs may involve sensory axons, motor axons, or a mixture of both.
The nerve is stimulated supramaximally, meaning that all the axons are electrically stimulated. In practice, this means that stimulator intensity (mA) is gradually increased while monitoring the size of the nerve response. When the response from the nerve gets no larger as the intensity continues to increase, this indicates that all the available axons have been activated and further increases in stimulation will activate no more axons. One should be aware, however, that increasing the stimulator substantially above this level will sometimes activate other nearby nerves, and responses from these other nerves can be recorded from the same recording electrodes (Fig. 4-2) through volume conduction; this is to be avoided.
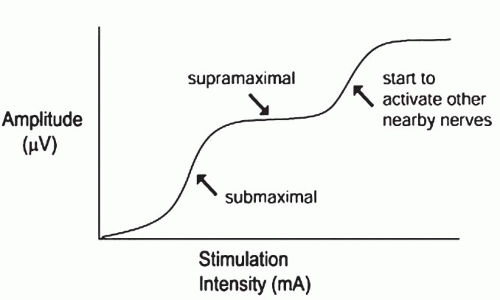 FIGURE 4-2. Amplitude of CMAP or SNAP as current is increased. Note that with very high levels of stimulation, amplitude will increase above supramaximal due to co-stimulation of nearby nerves. |
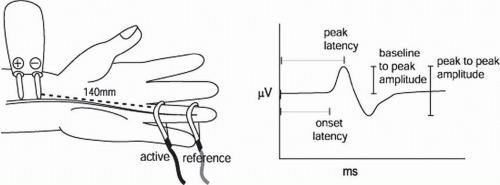 FIGURE 4-3. Example of an SNAP. |
One records from the same nerve at a specified or measured distance away from the stimulation and records the synchronized discharge of all the axons that have been activated (Fig. 4-3).
Several items are then measured from the nerve action potential. Most commonly, these include the latency and amplitude of the response. The latency can either be measured to the onset of the response or the peak. The advantage of measuring to the onset is that this represents the fastest conducted fibers, and it also depends less on the distance between active and reference electrodes. The advantage of measuring latency to the peak is that the peak is more reliably determined and less influenced by the sensitivity of the display or the amplifier. In either case, one should have reference (normal) data, which have been derived using the same technique. The amplitude or size of the response can be measured from baseline to peak or from peak to peak, again depending upon how the reference data were collected. Conduction velocity is in units of distance over time, in this case meters per second (m/s), which is the same as millimeters per millisecond (mm/ms). In the case of nerve action potentials, one can measure the distance between stimulation and recording and divide that by onset latency to obtain the velocity in meters per second.
Conceptually, one should consider what factors will influence the latency (or conduction velocity) and amplitude of sensory responses. The latency can be affected by a number of both physiologic and pathologic processes. Cold slows conduction velocity. This effect varies from nerve to nerve but generally is in the range of about 5% per °C. Conduction velocity also changes with age. Initially, children are born with conduction velocities about half of that of adults. As their nerves become more myelinated, these values approach normal values around age 3. Later, in the fifth and sixth decades, velocities start to slow again but not to the degree of that seen in infancy. For this reason, reference values are often adjusted for age.
With respect to pathological changes, loss of myelin will cause slowing of conduction velocity and increase in latencies. When demyelination becomes severe enough, however, conduction block will occur. One might think that such fibers could conduct slowly, as unmyelinated fibers do. However, since myelinated fibers do not typically have sodium channels underneath the areas of myelin, in the setting of intense demyelination, they cannot conduct as unmyelinated fibers do and so conduction is blocked.
The amplitude of the response is also affected by temperature. Cold tends to increase the amplitude of the response if the recording electrodes are over a cold area of the limb. This increase in size is because cold slows both the opening and, even more, the closing of sodium channels. As a result, more charge is exchanged across the axon plasma membrane and a greater size axon potential is elicited. This increased potential is then summated across many axons and the nerve action potential becomes enlarged. Amplitude is also dependent upon the distance of the electrodes from the generator source. The electrical potential amplitude falls off rapidly with distance as one moves further from the nerve. Thus, nerve conduction studies in which the response is close to the skin (such as recording from the fingers) usually generate a bigger amplitude response than when one is recording from proximal areas of the limb (where the nerves are located deeper).
Amplitude of the SNAP or CNAP can be thought of as representing the number of axons that are participating in the response. As one loses axons, the amplitude falls roughly in proportion to the degree of axon loss. In some cases, a reduction in amplitude can result from axon loss occurring between the stimulating and recording electrodes, in which case changes in the potentials would be seen immediately. However, SNAP amplitudes will also be reduced if sensory axons are affected anywhere from their cell bodies (which are located in the dorsal root ganglion or DRG) distally. In such cases, the axons undergo wallerian degeneration, and by 10 days after a proximal loss of sensory neurons or axons, the distal response will disappear due to axonal degeneration. On the other hand, if lesions are proximal to the DRG, then the distal axons will remain connected to their cell bodies in the DRG and will conduct normally, even if the patient has complete anesthesia.
Sensory nerve conduction studies can be most useful in diagnosis of lesions that are distal to the DRG, including plexopathies, entrapment neuropathies, and polyneuropathies. Sensory nerve conduction studies should usually be normal in the setting of radiculopathies (when injury is typically proximal to the DRG), central nervous system disorders, neuromuscular junction (NMJ) diseases, and myopathies (the latter two because recordings are obtained from nerve and not muscle).
One should be aware of several potential limitations in utilizing SNAP or CNAP studies. First, these can be technically challenging to obtain when responses are small, such as in
people with polyneuropathies, people with thick or calloused skin, or in older individuals. It can be challenging as well in unusual nerves that are not commonly studied. Hence, in less frequently studied nerves, one should interpret the absence of a response with caution and consider studying the contralateral limb as well as other sensory nerves. Electronic averaging of responses can be useful to elicit small amplitude responses that would otherwise be unnoticeable in the baseline electronic noise.
people with polyneuropathies, people with thick or calloused skin, or in older individuals. It can be challenging as well in unusual nerves that are not commonly studied. Hence, in less frequently studied nerves, one should interpret the absence of a response with caution and consider studying the contralateral limb as well as other sensory nerves. Electronic averaging of responses can be useful to elicit small amplitude responses that would otherwise be unnoticeable in the baseline electronic noise.
As mentioned above, temperature will have significant impact on sensory nerve conduction studies. One should consider that the limb might be cold if latencies are prolonged but amplitudes are large. In contrast, with disease states, latencies would be prolonged but amplitudes would usually be small. If one is unsure about whether temperature is having an influential effect, it is best to warm the limb. This author puts the limb into a plastic bag and then submerges it in a “cooler” or sink filled with warm water for 5 minutes. There are many other techniques, including warm air (e.g., from a hair dryer), infrared lamps, and other methods.
Compound Muscle Action Potentials and Motor Nerve Conduction Studies
Compound muscle action potentials (CMAPs) involve electrical stimulation of nerves and recording of the synchronized discharge of muscle fibers in the muscle supplied by the stimulated nerve.
As above, for sensory nerves, the nerve is stimulated supramaximally. As opposed to SNAP or CNAP, one now records not from axons but from muscle fibers at a specified or measured distance away from the stimulation, and records the synchronized discharge of all the muscle fibers that have been activated (Fig. 4-4).
As with SNAPs, several measurements are taken from the CMAP. The amplitude or size of the response is usually measured from baseline to peak. The latency is measured to the onset of the response. Conduction velocity is more complicated to calculate than for SNAPs. Because the latency includes not only conduction time along the nerve but also time for NMJ transmission (about 1 ms), one cannot simply divide the distance between stimulation and recording by the distal latency. Instead, we stimulate at two points along the nerve and take the difference in distance divided by the difference in latency to derive a conduction velocity (see Fig. 4-4).
The latency can be affected by the same factors as for SNAPs. Physiologically, cold and advancing age slow conduction velocity. With respect to pathological changes, demyelination will cause slowing of conduction velocity or increase in latencies. When demyelination becomes severe, conduction slowing and block will become evident.
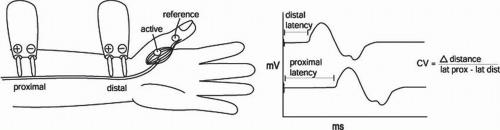 FIGURE 4-4. Example of a median CMAP. Conduction velocity is calculated by dividing the distance between the two stimulation sites by the difference in latencies. |
CMAP amplitude is dependent not only upon the number of axons that are participating in the response, but also on the integrity of the NMJ and the number and integrity of the muscle fibers supplied by the axons. So, while NMJ defects and myopathies will leave CNAPs and SNAPs unaffected, these lesions can produce a reduction in CMAP amplitude. The CMAP will fall roughly in proportion to the degree of motor axon loss (or, in the case of myopathies, muscle fiber loss). As opposed to the SNAP (where lesions anywhere from the DRG distally cause an amplitude reduction), a reduction in CMAP amplitude can result from lesions from the anterior horn cell (the motor neuron cell body) distally. By 7 days after a proximal loss of motor cell bodies or axons, the distal response will disappear due to axonal degeneration; this occurs earlier than for the SNAP due to earlier failure of the NMJ.
Motor nerve conduction studies can be most useful in diagnosis of lesions that affect the lower motor neuron or surrounding myelin. This includes significant spinal root lesions, plexopathies, entrapment neuropathies, and polyneuropathies.
Late Responses
While the typical motor and sensory conduction studies mentioned above rely on conduction distally in the limbs, there are several responses, known as late responses, which depend upon conduction proximally in the peripheral nervous system and thus occur with longer latency.
The F wave was so named because it was originally discovered in the foot (2). Physiologically, F waves depend upon conduction proximally to the motor neuron cell body. Since axons conduct in both directions, when motor axons are activated distally in the limb, they conduct both proximally and distally. For most axons, the impulse travels proximally, past the axon hillock, into the motor neuron and ends. But for a small number (about 3% to 5%), the depolarization traverses through the dendritic tree and comes back through the axon hillock after it is no longer refractory (about 1 ms). As a result, a small late response will be recorded from the muscle after enough time for a round trip from the stimulus site, back to the spinal cord and then back to muscle (Fig. 4-5). The reference (normal) values in humans are about 32 ms in the upper limbs and 55 ms in the lower limbs (3).
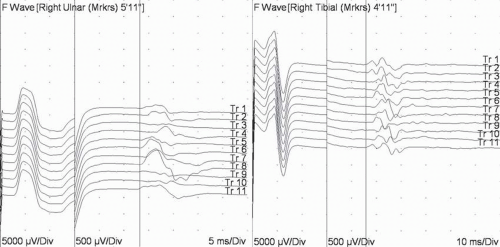 FIGURE 4-5. Examples of F waves from an upper limb (left) and a lower limb (right). Note the variability of the response. (Note also sensitivity is different on the left of the sweep than on the right.) |
The technique is similar to motor conduction studies but has several important differences (4). The sweep speed needs to be slow enough to capture the F waves (50 ms analysis time in the upper limb and 100 ms in the lower limb). Since F waves are smaller than CMAPs, sensitivity needs to be sufficient to observe these responses (200 to 500 μV/div); some EMG instruments have split screens with the first part of the sweep (e.g., first 20 ms) at 2 to 5 mV/div to observe the CMAP and the later part of the sweep at 200 to 500 μV/div. Conventional teaching is that the cathode should be placed proximally along the nerve with the anode distally to avoid proximal anodal block, but this probably makes little or no difference (5) as long as the cathode is placed at the same location. Conventional teaching is also that the stimulation should be supramaximal, but latencies are similar even with submaximal stimulation (6).
Since F waves are derived from different populations of motor neurons for each response, the latency and appearance vary with each stimulation. Thus, measuring latency is more complicated than for a CMAP or SNAP, where the latency is the same with each stimulation. Because of this variability, one must acquire multiple F waves (typically 10 to 20) and take measurements that consider this variability. Most commonly, one obtains either the minimal latency (the shortest latency of multiple responses) or the mean latency (the average latency). The minimal latency is easiest to measure and is most commonly used. However, the minimal latency will depend upon the number of responses obtained. As more responses are recorded, the chances of finding a shorter latency response increase and the minimal latency will drop. The mean latency, on the other hand, is more time-consuming to measure since one must mark all the F-wave responses obtained. But mean values are less dependent upon sample size, and the measure will be more stable, making it a more reliable parameter.
There are other latency measures that are also sometimes used. Maximal latency reflects the speed of conduction of the slower motor axons. Chronodispersion is measured as the difference between the shortest and longest F-wave latencies. Greater chronodispersion may indicate selective slowing of some motor axons more than others and can be expected in acquired demyelinating neuropathies (2).
Not every motor nerve stimulation results in an F wave. Persistence or penetrance is measured as the percentage of stimulations that produce an F wave. Persistence is altered by many factors (including activity level), and generally, low persistence is not considered a diagnostic finding (7). Absence of F waves, however, is considered abnormal.
Amplitude of the F wave can be measured, usually as a percentage of the corresponding CMAP. This, however, is also variable and is usually not used as a diagnostic finding.
As above, there are many measures one can obtain from F waves. A problem is that each measure has a false-positive rate which, if the reference (normal) values were analyzed optimally, is about 2.5%. As one performs more comparisons with reference (normal) data, each comparison roughly adds 2.5% to the false-positive rate. Thus, if one were to analyze minimal latency, mean latency, chronodispersion, persistence, and amplitude, there is roughly a 12.5% chance that at least one of these five measures would be “abnormal” in the healthy population. Consequently, it is this author’s preference to only measure the mean or minimal latency or indicate if the response is absent.
Although F waves will be abnormal in many clinical settings, they provide unique information in relatively few diagnoses. Probably the most useful setting for F waves is when evaluating for acquired demyelinating polyneuropathies, such as acute inflammatory demyelinating polyradiculoneuropathy (AIDP or Guillain-Barre syndrome) (8). In these disorders, the most proximal and distal ends of the peripheral nervous system (i.e., roots and distal axons) are affected first. As a result, one of the earliest findings on nerve conduction studies is absence of F waves or prolongation of F-wave latencies, presumably reflecting slowing or conduction block at the root level. This may be the only finding in some cases.
F waves also provide unique information regarding proximal conduction in early traumatic nerve lesions and will demonstrate changes before distal axon wallerian degeneration occurs and before changes appear on needle EMG. There is some evidence that F waves may change after walking in lumbar spinal stenosis (9), though it is unclear how F waves should contribute to the diagnosis of lumbar spinal stenosis (10).
There are a number of limitations to be aware of when employing F waves for diagnostic evaluation. First, since they involve only a few of the total pool of motor axons, lesions affecting only some of the axons can be missed. For instance, F waves are generally not helpful for detecting radiculopathies (11), although some authors have reported frequent abnormalities (12). Since most muscles have more than one root supplying them, normal F waves can be obtained via the other root supply, thereby bypassing slowing via the involved root. Another limitation is that F waves measure conduction over a long distance. This limits their ability to detect focal pathology that causes only focal slowing across a short segment of nerve, which is then diluted by much longer segments of normal nerve conduction. In addition to the above, F waves are generally applicable to only distal limb muscles. In proximal muscles, the F-wave latency is so short (it does not have far to go to the spinal cord and back) that the F wave is buried in the CMAP response.
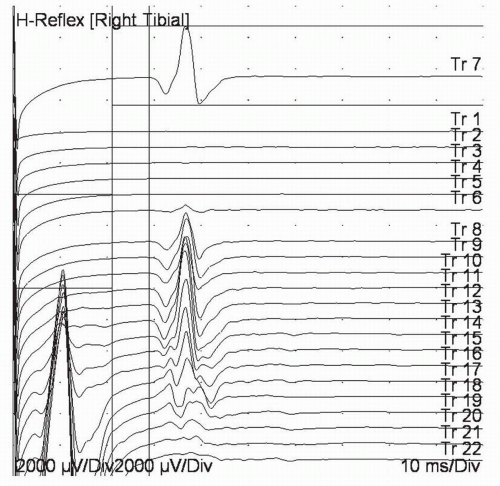 FIGURE 4-6. Tibial H wave recorded as stimulus intensity is increased (as trace number increases). Trace 7 at the top of the screen has been moved to the top for measurement. Note that as stimulus is increased, an H wave appears. With further increase in intensity, the H wave becomes smaller and the M wave (CMAP) grows larger. |
H waves (named after Dr. Hoffman) utilize a pathway similar to a muscle stretch reflex (2). In a muscle stretch reflex, the receptors in the muscle spindle are activated by sudden stretch of the muscle. This sends a wave of depolarization proximally up the large-diameter Ia sensory fibers to the spinal cord. At the spinal cord, there is a monosynaptic reflex to the α-motor neuron, and the descending motor axons are activated. H waves use a similar pathway, but the wave of depolarization starts at the Ia afferent fiber instead of the muscle spindle (13).
In normal healthy adults, H waves are easily elicited primarily in the soleus and the flexor carpi radialis muscles. Young children have H waves in many more muscles (before descending inhibitory motor pathways are fully developed). Patients with upper motor neuron lesions will also have easily elicited H waves in many other muscles (14).
When recording H waves, the sweep speed needs to be slow enough to capture the response (50 ms analysis times in the upper limb and 100 ms in the lower limb).
Sensitivity needs to be sufficient to observe these responses although H waves are typically larger than F waves. Similar to F waves, conventional teaching is that the cathode should be placed proximally along the nerve with the anode distally. However, one often uses less current if one stimulates through the knee, with a small 1-cm disc electrode over the tibial nerve (midway between the hamstring tendons at the popliteal fossa) and a 3-cm electrode (the type usually used as a ground electrode) anteriorly over the patella (15). Stimulation is best accomplished with long duration pulses (0.5 to 1.0 ms), since these preferentially activate the large Ia afferent nerve fibers that initiate the reflex arc (16). Stimulation should be at a level that produces the largest H wave, which is usually submaximal for the M wave (Fig. 4-6). Higher levels of stimulation are thought to inhibit H waves at the spinal level (2).
Latency is measured to the onset of the response—that is, the first departure from the baseline. Since the perceived onset will depend upon the display sensitivity (17), it is usually preferable to use a consistent display gain for measurement—ideally, the same used for one’s reference values (e.g., 500 μV per division). Latencies will depend upon height or leg length and most reference values take this into account. Side-to-side latency differences are more sensitive to detecting abnormalities than comparing a measured latency to reference values. A side-to-side latency difference exceeding 1.2 ms is likely abnormal for the tibial H wave (18). Amplitude of the H wave is variable and is also best compared to the other side. When the smaller response is less than 40% of the larger response, this is likely abnormal (18).
Although H waves will be abnormal in many clinical settings, they provide unique information in relatively few diagnoses. Probably the most useful setting for H waves is when evaluating for S1 radiculopathies. H waves are likely more sensitive for detecting S1 radiculopathies than is needle EMG (19). It is postulated that H waves can detect demyelination or sensory axon loss, while needle EMG only detects motor axon loss. H waves, however, provide less information about acuity or chronicity than needle EMG; they usually disappear early (or immediately) after onset of a radiculopathy and often never return. Obtaining and measuring H waves in the upper limbs are more challenging. They are somewhat more difficult to elicit than in the lower limb, and the onset is often obscured by the preceding M wave.
The A wave is another response that is sometimes noted during studies in which one is trying to record late responses. The A wave is a stable, small response that is the same with every stimulation (Fig. 4-7). A waves can appear with either submaximal or supramaximal stimulation. The proposed physiology is activation of motor axons due to either axon branching (usually seen with submaximal stimulation) or ephaptic transmission (seen with supramaximal stimulation) (Fig. 4-8). These are usually considered abnormal, though they are sometimes seen in the tibial nerve of healthy individuals (20). When seen with only submaximal stimulation, they may represent chronic nerve lesions with subsequent reinnervation and axon branching (21). A waves elicited with supramaximal stimulation are likely due to ephaptic transmission. These are sometimes seen in acute demyelinating polyneuropathies (e.g., AIDP) since demyelination creates an ideal setting for ephaptic transmission (see Fig. 4-8).
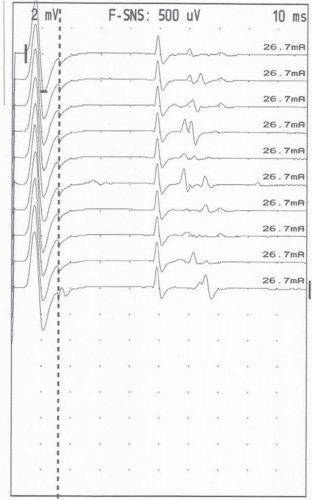 FIGURE 4-7. A wave. |
Needle EMG
Needle EMG is a very sensitive and useful technique for detecting pathology in the lower motor neuron, NMJ, and muscle. Key to an understanding of needle EMG is a familiarity with the concept of the motor unit (Fig. 4-9). The motor unit consists of the motor neuron cell body (which, for the limbs, resides in the anterior horns of the spinal cord), the axon, and all the muscle fibers the axon supplies. The number of muscle fibers innervated per axon varies widely. For small muscles that control very fine movements (e.g., extraocular muscles or laryngeal muscles), there may be only 4 to 6 muscle fibers per axon. In large muscles for which force is more important than fine control, such as the quadriceps, there are 500 or more muscle fibers per axon.
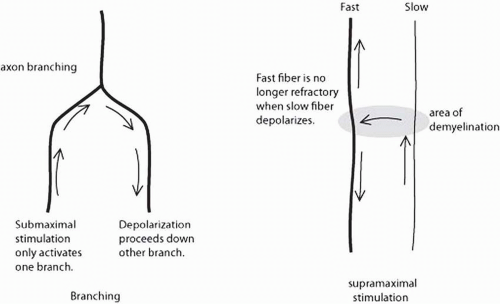 FIGURE 4-8. Potential A-wave mechanisms. On the left, branched fibers can produce a consistent late response if one branch is stimulated, but the other is not (i.e., submaximal stimulation). On the right, demyelination can produce A waves by ephaptic transmission. When stimulation occurs distally, the faster fiber has already depolarized in the site of demyelination by the time the slower fiber reaches the area and direct activation of the faster fiber can occur. |
In healthy individuals, if one puts a recording needle into a limb at rest, it is usually electrically quiet. When the individual gives a small voluntary contraction and activates the motor unit, a motor unit action potential (MUAP) can be recorded from the muscle (see Fig. 4-9). The MUAP represents the synchronous discharge of all the muscle fibers supplied by the axon. As more force is produced, these MUAPs fire more rapidly, and additional motor units are recruited until there are many MUAPs firing rapidly and none can be individually distinguished.
There are a number of potentials that can be recorded at rest (spontaneous potentials), some in healthy individuals, but most representing disease processes. When moving the needle electrode through the muscle, there is normally a brief electrical discharge from the muscle, known as insertional activity. This is usually high frequency and brief, usually lasting less than 300 ms (depending to some extent on the examiner). Insertional activity can be prolonged, with discharges persisting longer after needle movement than normal. Prolonged or increased insertional activity can be seen in early denervation, in some myopathies, and in some inherited syndromes (22) but is usually not considered diagnostic when seen in isolation. Reduced insertional activity is measured along a different dimension than increased insertional activity (which is measured in time). Reduced insertional activity refers to reduced amplitude or absence of the burst of muscle fiber discharges when moving the needle through muscle. Reduced insertional activity is seen when there are no resting muscle membrane potentials in the area of study, such as muscle fibrosis (long-term denervation), muscle necrosis (e.g., compartment syndrome) (23), or if it is not in muscle (but, for instance, in adipose tissue).
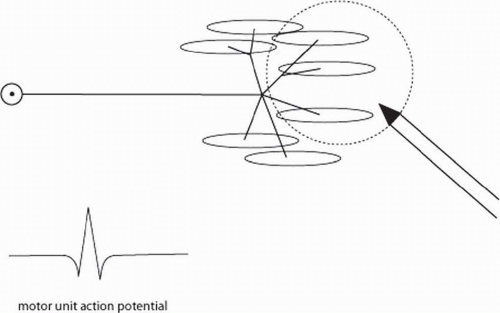 FIGURE 4-9. The motor unit. Each axon supplies from a few to hundreds of muscle fibers. The MUAP is recorded from muscle fibers of the motor unit that are within the recording volume of the electrode. |
In healthy individuals, one can easily enter the motor point of the muscle where there are abundant NMJs. When this occurs, one records endplate noise and endplate spikes. Endplate noise consists of small irregular potentials that sound (when played over a loudspeaker) like the sound of a seashell held up to one’s ear. They result from spontaneously released vesicles of acetylcholine (miniature endplate potentials or MEPPs). Endplate spikes are very brief (<5 ms duration) spikes with initially negative (upward) deflections, and they create a sputtering irregular sound similar to fat in a frying pan. They result from MEPPs summating to form localized endplate potentials (EPPs). These are normal findings, but one should pull back the needle electrode slightly and move to a different area for two reasons. These are typically painful areas to explore, and the endplate spikes, when viewed from a distance, can take on an initial positivity and look like fibrillation potentials.
Fibrillation potentials and positive sharp waves represent abnormal spontaneous single muscle fiber discharges and are frequently seen in lesions of the lower motor neuron or muscle
(24). They both represent single muscle fiber discharges and are seen in mostly the same disorders. Fibrillation potentials (Fig. 4-10) are short duration (1 to 5 ms), regularly firing spikes with an initial positivity (downward deflection). They are likely recorded by the needle electrode from outside the muscle fiber. Positive sharp waves (see Fig. 4-10) have a sharp initially positive (downward) deflection but have a duration of 10 to 30 ms, and they fire regularly. It is suspected that positive sharp waves are recorded from pierced or injured muscle fibers.
(24). They both represent single muscle fiber discharges and are seen in mostly the same disorders. Fibrillation potentials (Fig. 4-10) are short duration (1 to 5 ms), regularly firing spikes with an initial positivity (downward deflection). They are likely recorded by the needle electrode from outside the muscle fiber. Positive sharp waves (see Fig. 4-10) have a sharp initially positive (downward) deflection but have a duration of 10 to 30 ms, and they fire regularly. It is suspected that positive sharp waves are recorded from pierced or injured muscle fibers.
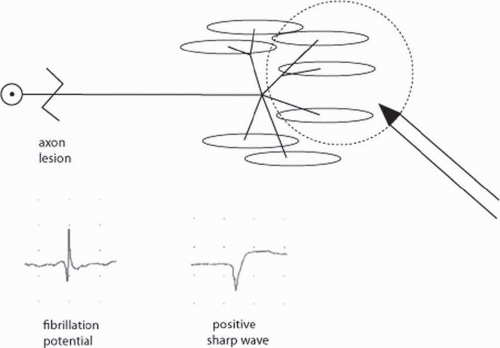 FIGURE 4-10. After axon loss, fibrillations and positive sharp waves are recorded. These represent abnormal, spontaneous single muscle fiber discharges. |
Both fibrillation potentials and positive sharp waves are seen in a variety of disorders and neither is specific to denervation (hence one should not use the term denervation potentials). They are commonly seen in disorders of motor neurons, motor axons, the NMJ (presynaptic more commonly than postsynaptic), and muscle (inflammatory myopathies and dystrophies more commonly). Upper motor neuron lesions (e.g., spinal cord injury, stroke, and traumatic brain injury) can also produce fibrillation potentials and positive sharp waves in weak muscles, though less commonly than lower motor neuron lesions do.
When fibrillation potentials and positive sharp waves are recorded, one usually grades them on a 1+ to 4+ scale (see table). The examiner should remember that this is an ordinal scale but not a ratio or interval scale. Thus, we know that a grading of 2+ represents more axon loss than 1+, but not necessarily twice as much. Moreover, a finding of 4+ fibrillations does not mean that all axons have been lost—this can be seen in very partial axon loss (25). Some authors have reported that positive sharp waves and fibrillations can be seen in normal parapsinal and foot muscles (26, 27, 28), but other authors have not found this to be common (29).
Fibrillation and Positive Sharp Wave Grading | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Complex repetitive discharges (CRDs) can be seen in a variety of chronic neuropathic and myopathic conditions. It is believed that CRDs result from abnormal muscle membranes that allow ephaptic transmission (i.e., a depolarizing muscle fiber activates an adjacent muscle fiber via local currents, not via any synapse). A pacer muscle cell fires regularly, thereby activating adjacent muscle fibers, which then fire in a sustained rhythmic pattern. CRDs are constant iterative discharges that appear and disappear suddenly without much variation in firing rate or amplitude. They sound like steadily operating machinery. They are not usually considered normal findings but suggest chronic axon or muscle fiber loss. CRDs are most commonly seen in chronic neuropathic and myopathic conditions but are not specific to either one. Some authors report that they can be found normally in select muscles such as the iliopsoas (30). If they are found in a myotomal distribution, they can be considered diagnostic of radiculopathy (11).
Fasciculation potentials represent spontaneous discharges of an entire motor unit or a large part of a motor unit. They often originate in the anterior horn cell but can also originate distally along the axon or a branch of the axon (31). Fasciculation potentials are recognized by their appearance and rhythm of firing. Individually, they look just like a single motor unit. However, they can be distinguished from a voluntary motor unit by their random discharge rate. These discharges are also spontaneous and not under voluntary control. To observe fasciculation potentials, one must often wait with the needle quietly in the muscles without moving the needle for a minute or more. Altering the sweep speed to a slower sweep (e.g., 100 ms per division) is often helpful. Fasciculations otherwise could be easily missed when one is rapidly looking through a muscle on a faster sweep speed.
Fasciculation potentials can be seen in a variety of disorders. Most commonly, one sees benign fasciculations. When asked, approximately 50% of people report fasciculations in their calves, worse with activity and sometimes increased with the use of caffeine (32). Fasciculation potentials are also seen in thyrotoxicosis and exposure to anticholinesterase medications. They can be seen in radiculopathies and chronic neuropathies as well. Perhaps the most worrisome disease in which fasciculations can be seen is motor neuron disease. Amyotrophic lateral sclerosis (ALS), progressive bulbar palsy (PBP), and other variants of motor neuron disease can all present with fasciculation potentials. The primary way
in which benign fasciculations can be distinguished from those associated with disease is by the “company they keep.” In motor neuron and other progressive disease processes, one will often see fibrillation potentials, positive sharp waves, and large amplitude long duration motor unit potentials, along with the fasciculation potentials. Benign fasciculations do not present with these other findings. When comparing fasciculations associated with motor neuron disease to those in benign fasciculations, there are some other minor differences, but these are not sufficient to be diagnostic by themselves. Fasciculations associated with motor neuron disease tend to be less frequent in firing rate, and they tend to be larger in amplitude, polyphasic, and longer duration (reflecting the concurrent ongoing reinnervation).
in which benign fasciculations can be distinguished from those associated with disease is by the “company they keep.” In motor neuron and other progressive disease processes, one will often see fibrillation potentials, positive sharp waves, and large amplitude long duration motor unit potentials, along with the fasciculation potentials. Benign fasciculations do not present with these other findings. When comparing fasciculations associated with motor neuron disease to those in benign fasciculations, there are some other minor differences, but these are not sufficient to be diagnostic by themselves. Fasciculations associated with motor neuron disease tend to be less frequent in firing rate, and they tend to be larger in amplitude, polyphasic, and longer duration (reflecting the concurrent ongoing reinnervation).
Myokymia is often thought of as a grouped fasciculation. Myokymic discharges represent groups of motor units firing in a burst pattern (usually with a regular burst rate) (33). These discharges, when played over the EMG instrument’s loudspeaker, are often described as sounding like marching soldiers. However, the bursts can be quite different in both their duration and discharge rate making recognition sometimes difficult. Myokymia is distinguished from CRDs because of the bursting nature rather than a single on and off with constant firing as seen in CRDs. Myokymic discharges are also distinguished from myotonic discharges because the former do not change in firing frequency or amplitude. Myokymia can be seen in a variety of conditions and is usually thought of separately for facial myokymia and limb myokymia. In facial myokymia, one often thinks of multiple sclerosis, pontine gliomas, and other brainstem disorders. In the limb, myokymia can be seen in some chronic compressions such as radiculopathies and entrapment neuropathies.
It is probably most important to remember that limb myokymia can occur in the presence of radiation plexopathy (34). This is useful when patients have had radiation for breast cancer, Hodgkin’s lymphoma, or other malignant tumors. Occasionally, these patients will present with a later-onset plexopathy, and the diagnostic question is whether the plexopathy represents tumor invasion or delayed-onset radiation plexopathy. Tumor invasion in the brachial plexus tends to present with pain, lower trunk lesions, and a Horner’s syndrome. Radiation plexitis presents with upper trunk lesions, paresthesias, and myokymia. Thus, myokymia argues for a diagnosis of radiation plexopathy in these cases.
Myotonia is a discharge that originates in single muscle fibers. It is believed to be due to abnormal chloride conductance across the muscle cell membrane. These discharges are noted by their unique waxing and waning quality, both in frequency and in amplitude. Because the firing frequency changes, the pitch produced over the loudspeakers changes as well, and one hears sounds often described as a “dive bomber” or “revving motorcycle.” The amplitude of the response also changes over time making the sound louder and softer during the discharge pattern. This can be distinguished from CRD because of its changing nature (CRDs are noticeably constant in firing frequency and amplitude).
Myotonia can be seen in a variety of muscle diseases including myotonic dystrophy, myotonia congenita, paramyotonia, and other disorders (35). Myotonic dystrophy is an unusual myopathy in that distal muscles are affected more than proximal muscles, and the same is true for where one might record these discharges. Distal hand muscles (e.g., first dorsal interosseous [FDI]) are often most affected. Myotonia is often worsened by cold and can be brought out by icing the distal part of the limb before recording. Myotonic discharges are also reduced immediately after exercise. Since myotonia originates from muscle, it is not expected in patients with neuropathic disease.
A possible variant of myotonia is the syndrome of diffusely abnormal increased insertional activity, also known as “EMG disease.” This disorder is an autosomally dominant inherited syndrome and presents with increased insertional activity and persistent positive sharp waves in essentially all muscles tested in the body (22). One can tell, however, that the patient does not have a true neuropathic disease because there are no fibrillation potentials, motor unit potentials are normal in morphology and size, and recruitment is normal. Because of the diffuse positive sharp waves, these patients can often be erroneously diagnosed with motor neuron disease or other serious disorders. It is difficult to know whether this syndrome produces symptoms or not, since those coming to the EMG laboratory are usually selected for some symptoms to start with. However, relatives of the patient with this disorder are often asymptomatic.
Motor Unit Analysis
Analysis of MUAPs yields important information about the integrity of the motor unit and whether there are changes in axons, muscle fibers, or the NMJ. The MUAP, as mentioned earlier, represents the synchronous discharge of all the muscle fibers supplied by a single motor neuron. Duration of the MUAP is largely influenced by the size of the motor unit territory, that is, the number of muscle fibers supplied by the single axon that are within the recording area of the needle electrode. Duration of the MUAP is not markedly affected by proximity of the depolarizing muscle fibers to the needle electrode.
Amplitude of the MUAP is also related to motor unit territory but is more influenced by the proximity of the discharging muscle fibers to the recording electrode. When the needle is close to the depolarizing muscle fibers, the amplitude is much larger than when it is some distance away. Thus, motor unit duration is a more reliable indicator of motor unit territory than is amplitude. There are reference values established for concentric electrodes for most of the major muscles in the body (36). These vary according to muscle and age. More proximal muscles, and especially bulbar muscles, have shorter duration MUAPs. MUAP duration tends to increase with age. There are not well-established reference values for monopolar electrodes. Thus, when performing quantitative MUAP analysis, one should use a concentric electrode.
Polyphasicity is noted when there are more than five phases of an MUAP. A phase represents a change in direction that crosses the baseline. Thus, one can calculate the number of
phases as the number of baseline crossings plus one. Changes in the direction that do not extend across the baseline are not counted as a phase. The presence of polyphasicity alone is usually not diagnostic, since most normal muscles have a small percentage (approximately 20%) of polyphasic MUAPs. However, polyphasic, long duration, large amplitude MUAPs are usually seen in neuropathies, and polyphasic, short duration, small amplitude MUAPs are typically seen in myopathic conditions.
phases as the number of baseline crossings plus one. Changes in the direction that do not extend across the baseline are not counted as a phase. The presence of polyphasicity alone is usually not diagnostic, since most normal muscles have a small percentage (approximately 20%) of polyphasic MUAPs. However, polyphasic, long duration, large amplitude MUAPs are usually seen in neuropathies, and polyphasic, short duration, small amplitude MUAPs are typically seen in myopathic conditions.
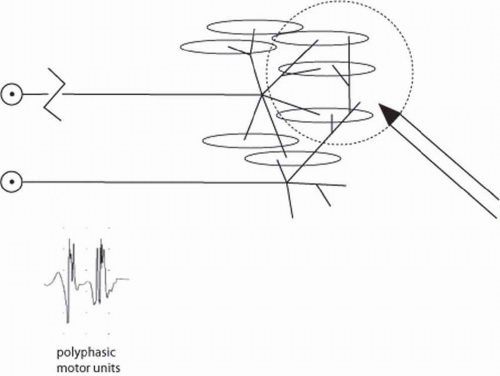 FIGURE 4-11. After partial axon loss and reinnervation by axonal sprouting, intact axons send new sprouts out to denervated muscle fibers. When the intact axon then fires, it is larger, polyphasic, and longer in duration. |
In neuropathic conditions, two types of reinnervation may be noted that change the MUAP morphology. When incomplete nerve injuries are present, some axons are spared, and others undergo wallerian degeneration. In this case, the remaining axons send distal sprouts to reinnervate the denervated muscle fibers. These sprouts are initially poorly myelinated and immature, which results in less synchronous discharge of muscle fibers than seen normally. When this distal axonal sprouting happens, one will observe polyphasic long-duration large-amplitude MUAPs (Fig. 4-11). As the new sprouts mature, the polyphasicity is reduced because the new sprouts become better myelinated and the muscle fibers fire synchronously again. Thus, late after reinnervation by axon sprouting, the examiner will see large amplitude long duration but not polyphasic MUAPs.
In cases of complete nerve transactions or complete axon loss, the picture is different. In these cases, there is no distal axonal sprouting because there are no viable distal axons. Rather, one is dependent upon axons regrowing from the site of injury down to the muscle. When these new axons first reach the denervated muscle, they innervate just a few muscle fibers. These new MUAPs (which used to be called nascent potentials) are typically short in duration, polyphasic, and small in amplitude because the axons have supplied only a few muscle fibers. As these axons continue to sprout, they become larger and longer duration and retain polyphasicity until the new sprouts mature several months after reinnervation.
Whether reinnervation occurs by distal axon sprouting or axon regrowth from the site of injury, recruitment will be reduced or discrete. Because there are fewer MUAPs, the existing motor units will fire more rapidly than normal, and there will be fewer MUAPs firing even with maximum contraction.
The model for myopathic changes in MUAPs can be thought of as random loss of muscle fibers within the motor unit (Fig. 4-12). In this case, since there are reduced numbers of muscle fibers, the MUAP duration and amplitude will be reduced. These MUAPs are polyphasic possibly due to less muscle fibers in the motor unit or possibly due to temporal dispersion along muscle fibers that conduct at different speeds in the setting of disease. In either case, in myopathies, one sees small-amplitude short-duration polyphasic MUAPs. These MUAPs are recruited early with many MUAPs appearing on the EMG screen with even small amounts of force generation. It is difficult to isolate a single MUAP in patients with the most severe myopathies.
NMJ disease can look, in many ways, just like a myopathy on needle EMG. Since the NMJs in a muscle do not all fire in these cases, there are less functional muscle fibers per motor unit, and the duration and amplitude of the MUAPs will be reduced. A feature commonly found in NMJ disease that is not seen in myopathy, however, is that of motor unit instability. Since the NMJs will fire variably and unreliably, the entire MUAP will vary in its size and morphology as muscle fibers fire or do not fire with each successive MUAP discharge. This motor unit instability can be seen in NMJ disease as well as in recent reinnervation, since in the latter case the NMJs are also not mature and reliable. To observe motor unit instability, it is most helpful to look at MUAPs with a trigger and delay line
so that the MUAP fires repetitively at the same location on the screen.
so that the MUAP fires repetitively at the same location on the screen.
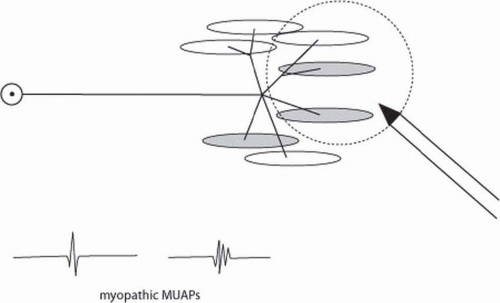 FIGURE 4-12. Myopathic MUAPs can be thought of as resulting from partial muscle fiber loss. With few muscle fibers in the MUAP, duration is shorter, amplitude is reduced, and there are more polyphasic MUAPs. The patient recruits more MUAPs for small levels of force since each MUAP creates less force than normal (early recruitment). |
While one typically evaluates MUAPs in a semiqualitative fashion, there are situations in which it is important to evaluate MUAPs more quantitatively. In the setting of possible myopathy, it is often very important to quantitatively measure MUAP duration to see whether it is shorter than reference values. Myopathies are sometimes more difficult to diagnose without quantitative analysis than neuropathic conditions, since one more frequently lacks the abnormal spontaneous activity or abnormal nerve conduction that are present in a neuropathic lesion.
To perform quantitative needle EMG, one must change the technical aspects of data collection in several ways. First, most reference values have been collected using concentric needle electrodes rather than monopolar needle electrodes. The concentric electrode has the advantage of a smaller recording volume and better reliability in duration measurements. Thus, one must use a concentric electrode. Normally, filters for needle EMG are set roughly from 20 Hz to 10 kHz. The low-frequency filter setting of 20 Hz is sufficient for capturing most electrophysiologic muscle potentials and reduces baseline wander, but it also distorts the low-frequency initial and terminal phases of the MUAP where departure from and return to baseline are gradual. For these reasons quantitative needle EMG requires that the low-frequency filter be reduced to 2 Hz, and multiple firings of individual MUAPs are averaged to present a clean single MUAP.
Multiple studies have been reported dating from the 1950s looking at how many MUAPs need be collected to obtain a reproducible mean MUAP duration for a particular muscle. These studies have shown that minimally 20 different MUAPs should be collected to obtain a reproducible value for mean MUAP duration, unless there are extremely abnormal MUAPs (37). Twenty different areas of the muscle should be studied, and this can usually be accomplished with only three to five different needle insertions through the skin. While quantitative needle EMG is commonly used in the setting of myopathy, generally only one or two muscles are studied this way. It is less common to use quantitative needle EMG in multiple muscles or in many neuropathic conditions where there are other hints of neuropathies.
After MUAPs are assessed, the electrodiagnostic medical consultant should then evaluate MUAP recruitment. Normally, when one provides a very small force, one will recruit a single motor unit firing slowly, typically about 5 Hz. As more force is exerted, this MUAP will speed up to 10 to 12 Hz and produce greater levels of force. However, as the firing rate reaches about 12 to 15 Hz (depending on the muscle), a second MUAP is then recruited to allow additional force production. In this way, muscle forces are generated both by increasing rates of MUAP firing and by increasing the number of MUAPs firing. Normally, at full contraction, there are many MUAPs firing rapidly and the baseline cannot be distinguished. There are generally three types of abnormalities of recruitment. Sometimes, patients have insufficient central drive from the upper motor neurons to generate high levels of MUAP firing. This could be due to pain, poor voluntary effort, or upper motor neuron lesions. In these cases, one sees less than the full number of MUAPs, but the ones that are present fire slowly due to reduced upper motor neuron drive. This is often termed central recruitment.
In other cases, there is sufficient upper motor neuron drive, but there are reduced numbers of motor units available to participate in force production. In these cases, the initial MUAPs fire more rapidly before a second MUAP is recruited because there are fewer motor units available to recruit. In extreme cases, there may only be one or two MUAPs firing very rapidly (up to 30 to 40 Hz) without any additional MUAPs present. This is typically termed reduced recruitment (or in cases where there are only very few MUAPs, discrete recruitment). It is distinguished from central recruitment in that the MUAP firing rate is quite fast.
A third abnormality one can see in MUAP recruitment is early recruitment. This is typically seen in myopathies or NMJ defects. With early recruitment, since each MUAP in a myopathy produces less force than normal, more motor units are recruited sooner or earlier than normal for a given level of force generation. The examiner asks the patient to produce a small amount of force and sees many MUAPs firing, more than expected for that level of force. In many cases of early recruitment, it is difficult to have a patient fire just one MUAP; it is usually either many or none. Thus, early recruitment represents many MUAPs firing for a low level of force production. Assessing early recruitment requires both measurement of force (whether qualitative or quantitative) and observation of MUAP firing patterns.
Repetitive Stimulation Studies
Repetitive stimulation studies are primarily useful for detection of NMJ abnormalities. To understand the underlying reasoning of how one performs NMJ studies, one needs to first review the physiology of NMJ, which is well described else-where (38).
Normally, when a motor axon is depolarized, the wave of polarization travels toward the axon terminal as a result of the propagation of opening and closing of sodium channels. When it reaches the terminal end of the motor axon near the end plate, voltage-gated calcium channels are activated. The opening of these channels allows an influx of calcium ions (Ca2+) into the presynaptic terminal. The opening of the voltage-gated and dependent calcium channels and the resulting calcium ion influx result in an increase of intracellular Ca2+ at the presynaptical terminal for about 100 to 200 ms before the Ca2+ is pumped out and the intra-axonal Ca2+ is returned to the resting concentration. This influx of calcium, however, allows the acetylcholine vesicles to fuse with the presynaptic membrane and release their quanta of acetylcholine into the extracellar space at the synapse.
At rest, the presynaptic terminal has occasional spontaneous release of quanta of acetylcholine that occur irregularly (38). When these quanta reach the postsynaptic terminal, they create MEPPs. However, when the presynaptic axon is excited or depolarized, the release of quanta is much greater. A high
concentration of acetylcholine is generated, which in turn activates the muscle fiber. In healthy individuals, the presynaptic terminal releases three to five times as much acetylcholine as is required to activate the postsynaptic membrane of the muscle fiber. This extra amount of acetylcholine release is known as the safety factor. While there is normally a large safety factor for the first discharge of a motorneuron, the amount of acetylcholine released drops with successive discharges when the nerve is activated at 2 to 3 Hz. However, because the safety factor is so large, there is still more than enough acetylcholine to activate the muscle fiber (Fig. 4-13).
concentration of acetylcholine is generated, which in turn activates the muscle fiber. In healthy individuals, the presynaptic terminal releases three to five times as much acetylcholine as is required to activate the postsynaptic membrane of the muscle fiber. This extra amount of acetylcholine release is known as the safety factor. While there is normally a large safety factor for the first discharge of a motorneuron, the amount of acetylcholine released drops with successive discharges when the nerve is activated at 2 to 3 Hz. However, because the safety factor is so large, there is still more than enough acetylcholine to activate the muscle fiber (Fig. 4-13).
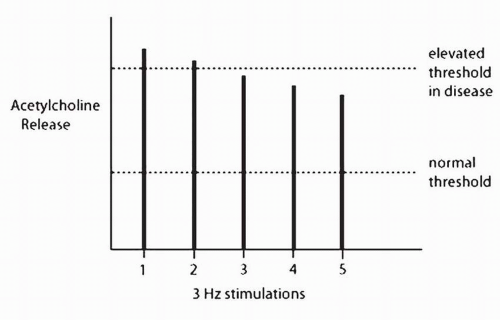 FIGURE 4-13. The amount of acetylcholine released with successive stimulations at 3 Hz drops. However, in the presence of a normal threshold for muscle fiber activation (normal safety factor), this drop is not noted. When the threshold is elevated and the safety factor reduced, some muscle fibers may not be activated. |
Once the acetylcholine is released into the synaptic cleft, it encounters acetylcholinesterase, the enzyme so named because it hydrolyzes acetylcholine molecules. This enzyme does not “know” whether the acetylcholine it is digesting has recently come from the presynaptic terminal or has already been used to activate the postsynaptic receptors. It digests the acetylcholine it encounters either way. Due to this enzymatic activity, it is estimated that only about half of the acetylcholine released from the presynaptic terminal makes it to the postsynaptic receptors. Once the acetylcholine reaches the postsynaptic receptors, it creates localized areas of membrane depolarization known as EPPs. These EPPs then summate to create a muscle membrane depolarization, and the muscle fiber discharges.
Exercise also has significant influences on NMJ physiology. Immediately after exercise, one sees postexercise potentiation or facilitation. In part, this is due to the fact that motor units fire at rates approaching 30 Hz with full voluntary contraction. At these rates, there is only roughly 33 ms between each axon depolarization, which is less than the time needed for the Ca2+ to be pumped out of the presynaptic terminal. Thus, with exercise, Ca2+ builds up in the presynaptic terminal, and this further facilitates release of acetylcholine. When the safety factor is intact, this is not noticed because there is already more than enough acetylcholine to fully depolarize the muscle fiber. However, when the safety factor is reduced in disease states, some NMJs do not fire, and postexercise potentiation will create a larger amplitude CMAP than preexercise. This is termed postexercise facilitation.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


