9 Dendritic Cells
Dendritic cells (DCs) are potent antigen-presenting cells (APCs) implicated in the induction of immunity and in the maintenance of tolerance. DCs represent a sparsely distributed population of bone marrow–derived mononuclear cells that are found in most tissues of the body. In the immature state, they are primed to capture antigens through expression of several receptors that enable recognition and acquisition of foreign and self-antigens.1 Upon encountering pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs), DCs undergo a process of “maturation,” involving changes in the DC phenotype, antigen acquisition capacity, migration, and an ability to traffic to draining lymph nodes, where they prime humoral and cellular immune responses.1 Under steady state conditions, DCs play an active role in maintaining tolerance to self-antigens. Although most autoimmune cells are deleted in the thymus through a process of negative selection, others must be tolerized through active and sustained mechanisms.
Dendritic Cell Subsets
In this chapter, we concentrate on human DCs with little additional reference to murine models. Readers are encouraged to seek additional information in several comprehensive reviews.2–6
Conventional DCs (cDCs)
Conventional DCs, or myeloid DCs, herein referred to as cDCs, comprise different subsets based on location, function, and phenotype (Table 9-1). cDCs are APCs with high phagocytic activity as immature cells and high antigen presentation as mature cells with the ability to secrete large quantities of cytokines.2 They express myeloid markers (CD11c, CD33, CD13) and high levels of major histocompatibility complex (MHC)-I, MHC-II, and co-stimulatory molecules (CD80, CD86). cDCs are present in the circulatory system and can be found in virtually every peripheral tissue, as well as in lymphoid organs. They are highly migratory cells that upon maturation express chemokine receptors, such as CCR7, allowing them to move from tissues to the subcapsular sinus and the T cell zone of lymphoid organs via afferent lymphatic and high endothelial venules. In lymph nodes, cDCs can regulate T cell responses both in steady state (to induce tolerance or anergy) and during infection (Figure 9-1). They are potent producers of IL-12 and prime naïve T cells toward a T helper (Th)1 profile.
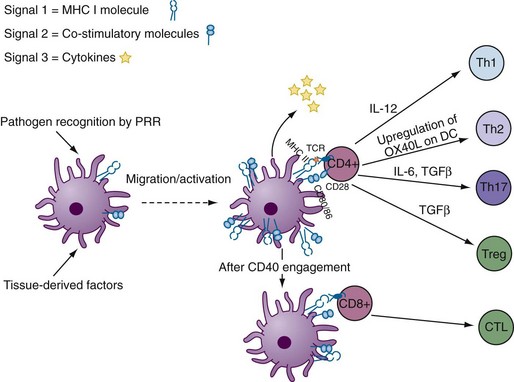
cDCs can be subcategorized into three components according to their localization: (1) peripheral tissue resident, such as skin; (2) secondary lymphoid organ resident; and (3) circulating blood cDC.5 Here we briefly describe some of the cDC subsets based on their localization. Understanding of DC subsets is critical not only in any future development of vaccines or treatments but also in understanding of autoimmune disease pathogenesis because each subset induces distinct responses.
Tissue Resident cDCs
Langerhans Cells (LCs)
LCs are located in the epidermis and have distinct markers, including CD1a and the C-type lectin receptor (CLR) langerin (CD207); LCs contain large granules called Birbeck granules. One of the pathways utilized by LCs to deliver antigen to T cells runs through langerin, which delivers antigen to Birbeck granules to be presented in a CD1a-restricted manner.7 The CD1 family of molecules is associated with presentation of glycolipids, and CD1a antigen presentation is independent of endosomal localization and acidification. LCs constitutively express E-cadherin, a homotypic adhesion molecule that anchors LCs to neighboring keratinocytes.8 Under steady state conditions, immature LCs migrate to the lymph node and induce T cell tolerance. During inflammation, migratory LCs upregulate MHC class II molecules, co-stimulatory molecules such as CD40, and CCR7, while expression of E-cadherin is downregulated, which facilitates LC disengagement from neighboring keratinocytes.8 In mice, LCs have been shown to be self-renewing in situ and are not replaced by bone marrow transfer in the steady state; data supporting their role as immunogenic APCs are conflicting.8
Dermal DCs
The CD14+ dermal DCs express a broad spectrum of C-type lectins, including DEC-205 (see Table 9-1). They have been shown to induce the differentiation of CD40-activated B cells into immunoglobulin (Ig)M-producing plasma cells through cytokine secretion (IL-12 and IL-6) and through direct cell-cell interaction.9 CD14+ DCs promote antibody responses through skewing of CD4+ T cells into T follicular helper (Tfh) cells, which induce naïve B cells to switch isotype and secrete large quantities of immunoglobulins.9
CD1a+ dermal DCs express blood DC antigen 1 (BDCA-1) and are present in the upper layer of the dermis. Phenotypically they are similar to LCs but lack expression of langerin and E-cadherin.5 On stimulation, CD1a+ DCs produce IL-15 and few proinflammatory cytokines, except IL-8, and chemokines also expressed by dermal CD14+ DCs.5 They can induce Th2 responses and cytotoxic T lymphocyte (CTL) responses, however with less potency than LCs or CD14+ DCs.
Lymphoid Tissue DCs
DC subsets can be identified in tonsils, lymph nodes, and spleen.6 Intestinal DCs can be found in the lamina propria, Peyer’s patches, solitary lymphoid tissue, and mesenteric lymph nodes, where they process and sample luminal and self-antigens for presentation.10 Here we briefly describe BDCA-3+ and CD103+ DCs.
BDCA-3+ DCs
BDCA-3+ (CD141) DCs are considered to be the counterpart of mouse CD8α+ DCs found in lymph nodes, based on functional and phenotypic analyses.11–13 BDCA-3+ DCs are found in lymph nodes, tonsils, bone marrow, and spleen, where they localize to T cell areas, but they are the rarest population of cDCs in blood. They express Toll-like receptor (TLR)3 and TLR8, and CLEC9A (DC natural killer [NK] lectin group receptor-1 [DNGR-1]), a C-type lectin that can act as a sensor of necrotic cells.11 These DCs phagocytose dead cells, process captured antigens, and cross-present them to CD8+ T cells. Because cross-presentation is essential for eradication of cancers, viruses, and other pathogens, BDCA-3+ DC cells present a promising target in vaccination strategies.14 The chemokine receptor XCR1 also seems to be selectively expressed by BDCA-3+ DCs.12 Activated NK cells and CD8+ T cells produce XCL1, a ligand for XCR1, at the site of infection and may serve as a signal for recruitment of BDCA-3+ DCs.15 On TLR stimulation, BDCA-3+ DCs produce IL-12 and IFN-β.14
CD103+ DCs
CD103+ DCs reside in the intestinal lymph node and efficiently induce expression of gut homing receptors CCR9 and α4β7 by responding T cells and B cells through a retinoic acid receptor-dependent mechanism.10 CD103+ DCs induce generation of regulatory T cells (Tregs) from naïve CD4+ T cells mediated through retinoic acid and transforming growth factor (TGF)-β.16 In Crohn’s disease, CD103+ DCs maintain their ability to induce CCR9 expression by T cells, similar to healthy control DCs in small intestine10; however, it remains to be determined whether they maintain their tolerogenic phenotype in Crohn’s disease.
Circulating DCs
BDCA-1+ (CD1c) DCs
BDCA-1+ DCs originate from bone marrow. They circulate in the blood and migrate to secondary lymphoid organs and peripheral tissues, such as spleen and tonsil, as resting interstitial DCs.6 They are hypothesized to be similar to mouse CD8α− DCs. BDCA-1+ DCs have a limited ability to produce IL-12 and CXCL10 (IFN-induced protein 10 [IP-10]) but produce IL-8, a chemoattractant for granulocytes, monocytes, and lymphocytes after stimulation with poly(I : C), a synthetic double-stranded RNA.13,17 They can also produce IL-1β upon poly(I : C) stimulation, which can be mediated through the retinoic acid inducible gene I (RIG-I), rather than TLR3.13 Mouse CD8α− DCs selectively express RIG-I.18 Similar to their mouse counterpart, they have the capacity to present antigens to CD4+ T cells but limited ability to cross-present.13
Monocyte-Derived Dendritic Cells (moDCs)
Monocytes are precursors to macrophages and can differentiate into DCs (moDCs). In the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4, monocytes acquire dendritic cell morphology, produce inflammatory cytokines, and skew naïve T cell responses in vitro. Although moDCs have not been identified in humans or mice in steady state, recent studies show that in mice, lipopolysaccharide (LPS) or LPS-expressing bacteria can mediate differentiation of monocytes to moDCs.19 These moDCs acquire DC morphology, localize to the T cell area via CCR7, and have similar antigen-presentation capacity as classic DCs, including cross-presentation.19 Hence, blood monocytes can be a reservoir of DCs in response to inflammatory situations.
Plasmacytoid DCs (pDCs)
pDCs have high capacity for type I IFN production. pDCs are found in peripheral blood, thymus, and many lymphoid tissues. pDCs display distinct plasma cell morphology, contain abundant endoplasmic reticulum, and express CD4 and high levels of IL-3αR (CD123) but lack myeloid antigens, including CD11c, and most lineage markers. pDCs secrete high amounts of IFN-α upon viral infection but no IL-12, and although they are primed to capture virus (e.g., flu), they generally display poor antigen capture and presentation capacity. Upon activation, pDCs show similar characteristics to cDCs (e.g., dendritic morphology, high expression of MHC class II molecules) but a limited capacity to prime naïve T cells.20 pDCs can prime naïve T cells toward the Th1 profile in IFN-α–dependent and IL-12–independent pathways upon viral/TLR stimulation21 but can also induce Tregs in an indoleamine-2,3,-dioxygenase (IDO)-dependent manner, possibly to contain immune activation that is triggered, as in SLE or chronic human immunodeficiency virus (HIV) infection.22 In contrast to cDCs, pDCs contain “lymphoid” mRNA transcripts for pre-T α chains, germline IgK, and Spi-B.
pDCs have been shown to accumulate in inflamed tissues, as in early stages of psoriasis, through their expression of ChemR23 and CXCR4.23
DENDRITIC CELL Development
The developmental origin of pDCs and cDCs is still debated because pDCs and cDCs can be derived from both the common lymphoid progenitor (CLP) and the common myeloid progenitor (CMP), suggesting that pDCs and cDCs may arise during hematopoiesis from progenitors with already distinct and restricted lineage potential.24 Most studies on the developmental origin of human DC subsets have used in vitro culture systems. DC precursors, similar to other hematopoietic cells, are continuously produced at a steady rate in a pathogen-independent manner from CD34+ hematopoietic stem cells (HSCs) within the bone marrow. DC development arises through several commitment steps in the bone marrow, from hematopoietic precursors, including macrophage/DC precursors (MDPs) and common DC precursors (CDPs). Proliferative CDPs then differentiate into DC precursors (pre-DC) and pDCs. Pre-DCs localize in bone marrow, blood, and lymphoid organs, where they give rise to immature DCs (imDCs) and mature DCs (mDCs). The pre-DC subset expresses several myeloid markers, including CD11b, CD11c, CD13, CD14, and CD33.
Maturation
In their resting state, imDCs are highly specialized in antigen uptake through a variety of receptors and mechanisms. Upon encountering a pathogen or other “activation stimuli,” DCs undergo phenotypic and functional changes referred to as maturation; this influences their “effector” function and antigen-presenting capability (see Figure 9-1).1
Receptor Upregulation
Maturation is accompanied by increased expression of chemokine receptors, adhesion molecules, and co-stimulatory molecules that allow DC migration toward secondary lymphoid organs, facilitating interaction with T cells. DCs upregulate CCR7, which directs cells to lymphoid tissue in response to chemokines CCL19 and CCL21 expressed in the T cell areas of lymph nodes. Adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) are also upregulated upon maturation and bind lymphocyte function–associated antigen-1 (LFA-1) on T cells.25 Mature DCs are also characterized by high-level expression of MHC and T cell co-stimulatory molecules with an enhanced ability to present antigen captured in the periphery to T cells. DCs express various members of the B7 superfamily, such as B7-1 (CD80) and B7-2 (CD86), which stimulate or inhibit T cell activation, respectively, upon binding T cell CD28 or CTLA-4.26 DCs constitutively express CD40, which is upregulated upon maturation and binds CD40L (CD154) expressed by activated CD4+ T cells, resulting in enhanced upregulation of MHC molecules and increased production of cytokines and DC survival.27
Antigen Acquisition and Recognition
Toll-Like Receptors (TLRs)
TLRs recognize a variety of PAMPs, such as bacterial, fungal, and nucleic acids (Table 9-2). They can be grouped into two subfamilies: TLRs expressed on cell surface (TLR1, -2, -4, -5, -6) sampling for the presence of bacterial, fungal, and protozoan components; and TLRs localized to specialized endosomal compartments (TLR3, -7, -8, and -9), detecting the presence of nucleic acids28 (see Chapter 18). Although most TLRs appear to function as homodimers, TLR2 forms heterodimers with TLR1 or TLR6, and TLR4 is complexed with MD-2. DC subsets show a heterogeneous TLR expression pattern (see Table 9-2).
Table 9-2 TLR Expressed by Dendritic Cells (DCs) and Their Ligands
| Conventional DCs | Plasmacytoid DCs | Ligands |
|---|---|---|
| TLR1 | TLR1 | Multiple triacyl lipopeptides |
| TLR2 | Peptidoglycan (Staphylococcus aureus), lipoproteins, and lipopeptides from several bacteria Glycophosphatidylinositol anchors from Trypanosoma cruzi Lipoaminomannan from Mycobacterium tuberculosis, zymosan (yeast) | |
| TLR3 | Double-stranded RNA (e.g., poly I : C) | |
| TLR4 | Lipopolysaccharide + MD-2, taxol, HSP60 (?), heparan sulfate (?), RSV, fibronectin | |
| TLR5 | Flagellin (Salmonella typhimurium, Listeria) | |
| TLR6 | TLR6 | ?/Undergoes dimerization with TLR2 |
| TLR7 | Imiquimod (Aldara), R848 (resiquimod), single-stranded RNA | |
| TLR8 | TLR8 | Imiquimod (Aldara), R848 (resiquimod), single-stranded RNA |
| TLR9 | CpG ODNs, DNA from bacteria and viruses, chromatin-IgG complexes | |
| TLR10 | ? |
CpG ODNs, CpG oligodeoxynucleotides; HSP, heat shock protein; Ig, immunoglobulin; RSV, respiratory syncytial virus; TLR, Toll-like receptor.
Activation of DCs in response to binding of TLR agonists is controlled by signaling through the Toll/IL-1 receptor (TIR) domain present in the cytoplasmic domain of TLR. Signaling downstream of the TIR domain is mediated through recruitment of myeloid differentiation factor 88 (MyD88) for all TLRs, except for TLR3. MyD88 recruitment is required for induction of nuclear factor κB (NFκB), which induces expression of proinflammatory cytokines such as IL-12, tumor necrosis factor (TNF), and IL-6. In pDCs, stimulation of TLR7 and TLR9 with nucleic acid induces IFN response factor 7 (IRF7) through MyD88 signaling, leading to IFN-α secretion.29
TLRs play an important role in induction of autoimmunity. Under steady state conditions, DC responses to extracellular self-nucleic acids are prevented by the endosomal seclusion of nucleic acid–recognizing TLRs. However, cellular proteins and chaperones can bind and present self-nucleic acids to DCs, causing inflammation. For example, LL37 (cathelicidin), an antimicrobial peptide produced by neutrophils and keratinocytes, is found in high levels in the skin lesions of psoriasis patients. LL37 forms aggregates with self-DNA and RNA from necrotic cells, protects them from degradation, and transports them into endosomal compartments of DC. In pDCs, RNA-LL37 and DNA-LL37 complexes activate TLR7 and TLR9, respectively, and trigger the secretion of IFN-α.30,31 In contrast, RNA-LL37 complexes in cDCs trigger TLR8 and induce secretion of proinflammatory cytokines.30 Similarly, high mobility group B1 (HMGB1) is released from necrotic cells and stimulates TLR2 and TLR4, mediating secretion of proinflammatory cytokines.28 Heat shock proteins (HSPs), such as HSP60, HSP70, gp96, and HSP22, are also reported to be recognized by TLR2 and TLR4.28
C-Type Lectin Receptors (CLRs)
DCs can also recognize, bind, and uptake specific carbohydrate moieties expressed by pathogens or self-tissues through CLRs such as the macrophage mannose receptor (MMR), DEC-205, DC-specific ICAM-3 grabbing nonintegrin (DC-SIGN), Dectin-1, Dectin-2, dendritic cell inhibitory receptor (DCIR), myeloid inhibitory C-type lectin-like receptor (MICL, CLEC12A), CLEC9A, and BDCA-2, all of which can function as endocytic receptors to internalize antigens for degradation or antigen processing and presentation.32,33
CLRs Modulate TLR Signaling
CLRs can positively and negatively modulate TLR responses. DC-SIGN recognizes mannose and fructose on pathogens such as HIV-1, measles virus, Mycobacterium tuberculosis, and Candida albicans. Modulation of TLR signaling by DC-SIGN is dependent on prior activation of NFκB by TLR.33 Acetylation of NFκB p65 mediated by DC-SIGN prolongs and increases transcriptional activation from the IL-8 and IL-10 promoters, leading to increased cytokine secretion.33
On the contrary, binding of Salp15, a protein antigen from the tick Ixodes scapularis, to DC-SIGN decreases TLR1-TLR2–dependent inflammatory cytokine production by decaying IL-6 and TNF mRNA while impairing nucleosome remodeling at the IL12a promoter.34
DC-SIGN has an essential role in DC–T cell interaction through ICAM-2 and ICAM-3, which are highly glycosylated structures. In RA synovium, the numbers of cells expressing DC-SIGN on macrophages and DC and ICAM-3 on naïve T cells were increased in the affected joints, with interaction leading to increased secretion of matrix metalloproteinases.35
BDCA-2 expressed by pDC pairs with FcεRγ, which contains an immunoreceptor tyrosine-based activation motif (ITAM). Activation of BDCA-2 by antibody ligation results in downregulation of TLR9-mediated IFN-α, IFN-β, TNF, and IL-6 in pDCs.36 HIV gp120 was also found to inhibit TLR9-mediated IFN-α through binding of BDCA-2 on pDCs.37 SLE patients have reduced expression of BDCA-2 on pDCs, and lack of negative regulation through BDCA-2 signaling may account for excessive IFN-α production on TLR stimulation.38
DCIR and MICL are the only known CLRs with immunoreceptor tyrosine-based inhibitory motifs (ITIMs) in their cytoplasmic tails, which recruits SH2-domain–containing protein tyrosine phosphatase 1 (SHP-1) or SHP-2 following antibody ligation.33 So far no ligands have been identified for DCIR and MICL. Activation of DCIR leads to internalization into an endosomal compartment, with TLR8 and TLR9 leading to the downregulation of TLR8-induced TNF and IL-12 by cDCs, or TLR9-induced IFN-α and TNF production by pDCs.33 Polymorphisms in the gene encoding DCIR have been associated with RA.39 In mice, DCIR deficiency leads to unrestricted expansion of DC numbers and development of autoimmune sialadenitis, enthesitis, and autoantibodies, including rheumatoid factor, anti-Ro and La, and antinuclear antibodies, implicating DCIR in the negative regulation of DC expansion or differentiation from bone marrow precursors.40
Similarly, antibody-mediated cross-linking of MICL expressed by cDCs leads to SHP-1 and SHP-2 recruitment and the suppression of TLR-induced IL-12 production.33
CLR Signaling Independent of TLR
Dectin-1 recognizes β-1-3 glucan expressed by fungal pathogens. Dectin-1 induces two different signaling pathways: activation by recruitment of spleen tyrosine kinase (SYK)-dependent activation of NFκB, and activation of RAF1, leading to phosphorylation of p65, similar to DC-SIGN and TLR cross-talk. Activation and modification of NFκB by dectin-1 leads to an increase in IL-6, IL-10, IL-1β, and IL-12p40 production and modulation of IL12p35 and IL-23p19 subunits, leading to bioactive IL-12p70 and IL-23, respectively, resulting in generation of Th1 and Th17 cells.33
Dectin-2, a CLR, can associate with Fcγ receptors (FcγRs) similar to BDCA-2. Unlike BDCA-2, Dectin-2 induces cytokine production through FcγRs. Dectin-2 recognizes fungal hyphae from C. albicans, Trichophyton rubrum, and Microsporum audouinii and recruits SYK to FcγR and induces production of TNF and IL-6.33
CLRs Enhance Antigen Presentation
The ability of CLRs to deliver antigen to different compartments for processing has been used for targeting in vaccine studies.2 It has been shown that antigen linked to DEC-205–specific antibodies resulted in enhanced antigen uptake and presentation by DCs to both CD4+ and CD8+ T cells.41 CLRs have a specific expression pattern, and some CLRs, such as DC-SIGN and Dectin-1, are expressed by several DC subsets, while expression of other CLRs is restricted to specific DC subsets (see Table 9-1). CLEC9A recognizes necrotic cells and on binding recruits Syk; in mice this activation is required for cross-presentation of necrotic cell–associated antigens by CLEC9A.42 BDCA-3+ cells in humans express CLEC9A and have been shown to cross-present viral antigens to CD8+ T cells and could potentially play a part in cross-presentation of self-antigen in autoimmune diseases.12,13
Because CLRs are involved in immune homeostasis, their dysregulation might contribute to the predisposition to autoimmune disease. This is supported by associations of single-nucleotide polymorphisms of CLR and key components of their signaling pathways with a number of autoimmune diseases, including CLEC16A with type 1 diabetes, multiple sclerosis, juvenile rheumatoid arthritis, and inflammatory bowel disease; DCIR with rheumatoid arthritis; and CARD9 with the spondyloarthropathies, ankylosing spondylitis, and inflammatory bowel disease.43–47
Retinoic Acid Inducible Gene I (RIG-I)-Like Receptors (RLRs)
RLRs are cytoplasmic PRRs, recognizing genomic or replication intermediate viral double-stranded RNA (dsRNA). The RLR family is composed of RIG-I, melanoma differentiation-associated gene 5 (MDA5), and LGP2. RIG-I and MDA5 recognize short dsRNA with 5′- triphosphate ends and long dsRNA, respectively. LGP2 functions as a positive regulator of both RIG-I and MDA5.28 The CARDs of RLR activate a signaling cascade, leading to expression of type I IFN genes via TRAF3 and IRF3/7. In addition, IPS-1 induces nuclear translocation of NFκB via cleavage of caspases 8 and 10.28
Nucleotide-Binding Oligomerization Domain (NOD)-Like Receptors (NLRs)
The NLR family of proteins consists of cytosolic, intracellular PRRs that recognize PAMPs and endogenous ligands, which induce a signaling cascade leading to activation of NFκB, or a cytoplasmic multiprotein complex known as the inflammasome, to produce inflammatory cytokine.48 NLR family proteins have a trimodular structure and contain the following domains: a central nucleotide-binding NOD domain, essential for self-oligomerization, and a C-terminal domain consisting of tandem leucine-rich repeats (LRRs), essential for sensing and recognition of PRR.48 Some proteins contain variable N-terminal domains. NOD1 contains a CARD domain, which activates transcription of proinflammatory mediators, whereas NACHT-LRR-PYD–containing protein 3 (NALP3) contains a pyrin domain (PYD), and the NLR family apoptosis inhibitory protein 5 (NAIP5) contains a baculovirus inhibitor of the apoptosis protein repeat (BIR) domain. PYD and BIR domains are components of the inflammasome that regulate caspase-1 activation, leading to the production of IL-1β.48
NOD1 and NOD2 sense various bacterial PAMPs in the cytoplasm. Upon PAMP recognition, they undergo self-oligomerization through the CARD domain and form a complex with CARD-containing IL-1β-converting-enzyme–associated kinase (CARDIAK). This induces inflammatory cytokines through activation of NFκB and mitogen-associated protein kinases (MAPKs).48
Pathogen infection of DCs results in the production of various cytokines of the IL-1 family, including IL-1β, IL-18, and IL-33 through TLR, NLR, and RLR. The production of these cytokines is dependent on PRR-mediated translation and transcriptional upregulation of pro-forms of these cytokines. Inflammasomes, activated by caspase-1, process cytosolic pro-IL-1β and pro-IL-18 to bioactive and secretory cytokines.48 Inflammasomes are categorized into three types: NALP3, IL-1β-converting-enzyme-protease-activating factor (IPAF), and NALP1 inflammasome. They are activated directly or indirectly by various ligands. The NALP3 inflammasome can be activated by exogenous ligands such as viral RNA and by host endogenous ligands, including monosodium urate (MSU) and adenosine triphosphate (ATP).48 Gout, an acute and chronic inflammation of joints, is associated with the deposition of MSU, which most likely induces inflammation through NALP3 inflammasomes.49
NLRs are also involved in various inflammatory and autoimmune diseases. NOD2 signaling has been shown to induce Th17 responses for clearance of bacteria through the selective induction of IL-23 and IL-1, which is abrogated in DCs from Crohn’s disease patients carrying a mutation in NOD2.50 ATG16L1, an autophagy-related gene, was also identified in Crohn’s disease. In mice, deletion of the ATG16L1 gene leads to hyperactivation of inflammasomes and subsequent secretion of bioactive IL-1β and IL-18, contributing to intestinal inflammation.28 Therefore inappropriate responses to bacterial pathogen by DCs may contribute to the pathogenesis of Crohn’s disease.
Apoptotic Cell Recognition Receptors
In addition to sampling for immunogenic antigens, DCs help in clearance of apoptotic cells in the periphery. Upon uptake of apoptotic cells or apoptotic microparticles, DCs maintain an immature phenotype and induce immunomodulatory effects. These DCs downregulate co-stimulatory molecules (CD80/86) and produce TGF-β, a cytokine necessary for differentiation of naïve T cells to Tregs.51 In contrast, uptake of necrotic cells leads to maturation and secretion of proinflammatory cytokines. Inability to clear apoptotic cells and consequent secondary necrosis have been implicated in the development of SLE. Recognition and engulfment of apoptotic cells involve multiple ligands: “eat me” signals, bridging molecules, and phagocytic receptors. One of the principal “eat me” signals is translocation of phosphatidylserine (PS) to the outer leaflet of the plasma membrane of apoptotic cells. PS can be recognized by multiple receptors, such as T cell immunoglobulin mucin (TIM-1), TIM-4, Stabilin-2, brain-specific angiogenesis inhibitor 1 (BAI1), and scavenger receptors (e.g., CD36, CD68, LOX-1). In addition, the β2-glycoprotein I (β2-GPI) receptor, αv integrins, and MER tyrosine kinase recognize PS through bridging molecules β2-glycoprotein I (β2-GPI), milk fat globular-EGF factor 8 protein (MEGF-8), and Gas6, respectively. CD36 and αvβ3 on DCs bind apoptotic cells via bridging molecule thrombospondin-1 (TSP-1); complement receptor 3 (CR3 [CD11b/CD18]) and CR4 (CD11c/CD18) via complement protein C3bi; and scavenger complex calreticulin-CD91 via surfactant proteins A (SP-A), SP-D, and C1q.52 CD14 and ATP-binding cassette transporter (ABC) 7 have also been implicated in recognition of apoptotic cells. In addition to providing “eat me” signals, apoptotic cells modify CD31, which prevents ingestion of viable cells.52
DCs can also phagocytose necrotic cells; LOX-1 binds to heat shock proteins (HSPs) and allows cross-presentation to T cells of antigens bound to HSP.53 Recently, variants of ITGAM (CD11b) were associated with SLE.54 Polymorphisms in C1q, a complement factor, have also been linked with SLE.55 Polymorphisms in C1q and CD11b could lead to decreased uptake of apoptotic cells and subsequent necrosis and proinflammatory responses.
A common feature of lupus patients is the presence of dying cells in both lymph nodes and inflamed tissues.56 It is not clear whether this is due to increased cell death or innate or acquired inability to clear apoptotic cells, which normally occurs very rapidly. Increased cell death might be the consequence of oxidative stress or infection, both of which are thought to trigger autoimmune disease. Regardless of the mechanism, increased cell death increases the presence of autoantigens available for presentation by DCs. Necrotic cells attract phagocytes by secreting hyaluronic and uric acid and high mobility group box 1 protein (HMGB1), which are known inducers of inflammasome and inflammation.57
Fc Receptors (FcRs)
Infected cells or pathogens coated with IgG activate FcγR-mediated clearance by antibody-dependent cell-mediated cytotoxicity (ADCC) or phagocytosis and/or indirectly through the release of cytokines. This is mediated by ITAM phosphorylation and subsequent activation of SYK and the initiation of the downstream signaling cascade resulting in cell activation.58 In contrast, inhibitory FcγRIIB has an ITIM domain in its cytoplasmic domain, resulting in inhibition of phagocytosis and cytokine secretion.
FcγRIIB also controls IC-mediated DC maturation.59 Low levels of IC can be identified in the serum of healthy individuals, thus FcγRIIB can prevent spontaneous DC maturation. In a small subset of well-controlled RA patients who were able to stop disease-modifying drugs, peripheral blood monocyte-derived DCs expressed high levels of FcγRIIB, signaling through which inhibited TLR4-mediated DC activation.60 In addition, ICs taken up by FcγRIIB are degraded inefficiently and are presented on the cell surface in native conformation, where they can interact with B cells.61 An FcγRIIB gene polymorphism has been identified in SLE and RA patients. DCs from RA patients with FcγRIIB variant showed an increase in IC-mediated inflammation in vitro.62
FcγRs also alter CR3-mediated signaling of phagocytosis upon co-stimulation in vitro. Co-stimulation of CR3 with FcγRI inhibits and FcγRIIA enhances CR3-mediated phagocytosis63—a potential mechanism for selective modulation of immunity.
Antigen Presentation
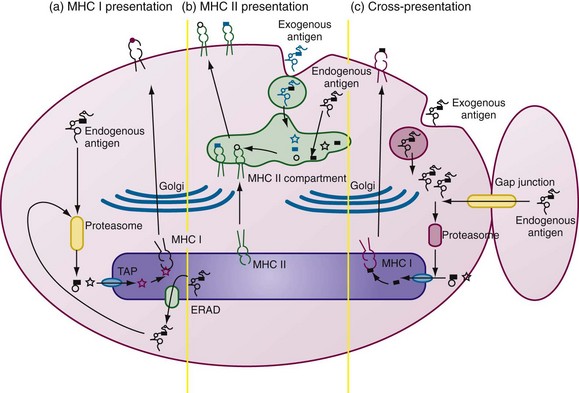
DC activation is tightly associated with processing and presentation of antigens by MHC class I and MHC class II, and lipid presentation by CD1 molecules (Figure 9-2).
MHC Class I Antigen Presentation
Antigenic peptides are predominantly generated from the ubiquitin-proteasome system of ubiquitinated nascent proteins, misfolded proteins (retrotranslocated from endoplasmic reticulum [ER] to the cytosol through the ER-associated degradation pathway [ERAD]), neosynthesized defective proteins, defective ribosomal products (DRiPs), and proteins expressed in the cytoplasm. IFN-γ strongly influences processing efficiency by inducing immunoproteasome formation and proteasome activator PA28 synthesis.64 DCs express immunoproteasomes containing the active site subunits LMP2, LMP7, and MECL-1, which enhance antigen processing and the generation of a different spectrum of peptides from the standard proteasome. Recently, intermediate immunoproteasomes were identified; they contain only one or two inducible catalytic subunits of immunoproteasome and confer different cleavage specificities. This broadens the repertoire of antigens presented to CD8+ T cells.65
The peptides are transported to the ER through the transporter associated with antigen processing (TAP), where long peptides are further trimmed by ER aminopeptidase-1 (ERAP-1) to 8-mer or 9-mer peptides for loading onto MHC class I molecules. The trimeric complex of MHC class I heavy chains, β2 microglobulin, and peptide allows for optimal folding, glycosylation, and delivery to the cell surface (see Figure 9-2).
Cross-Presentation
DCs can acquire antigens exogenously and process them for presentation by MHC class I molecules, referred to as cross-presentation.66,67 This property is atypical because MHC class I molecules exclusively present endogenous proteins in most cells. Cross-presentation requires peptides to gain access to cytosol, to be processed by proteasome, and to be transferred to the ER for loading onto MHC I class molecules (see Figure 9-2C). Although the precise mechanism of cross-presentation remains controversial, it is well established that DCs utilize this process to activate CD8+ T cells. DCs may acquire antigens in the form of apoptotic cells, necrotic cells, antibody-opsonized cells, immune complexes, heat shock proteins, and exosomes, and even by nibbling of live cells.52,66–69 Autophagosomes may be another source of peptides for MHC class I molecules. Autophagy clears ubiquitinated cytoplasmic protein aggregates.70 Activation of DCs through TLR or exposure to stress induces DC aggresome-like structures (DALIS) containing polyubiquitinated proteins; peptides derived from DALIS can also be presented by MHC class I molecules.71
MHC Class II Antigen Presentation
The MHC class II pathway is constitutively expressed only by APCs. MHC class II αβ heterodimers rely on a specialized type II transmembrane chaperone protein, the invariant chain (Ii), for stable assembly in the endoplasmic reticulum.72 Ii stabilizes the MHC II complex and contains an endosomal sorting and retention signal.72 MHC class II molecules are transported and concentrated in multivesicular and multilamellar late endosomal compartments called the MHC class II-containing compartment (MIIC).
Antigens are endocytosed and retained within phagosomes before fusing with lysosomes to form phagolysosomes. Concomitant TLR signals induce activation of the vacuolar proton pump that enhances lysosomal acidification and antigen proteolysis within phagolysosomes.73 Acidification of this compartment allows optimal activity of cathepsins. Cathepsin S degrades the cytoplasmic tail of Ii, leaving a short peptide, the MHC class II–associated invariant-chain peptide (CLIP), bound to the peptide-binding groove and thus protected from proteases. CLIP is replaced by an antigenic peptide in the endosome derived through the action of endosomal proteases on essentially any proteins accessing the endosome. The rate of dissociation of CLIP is too slow for quantitative peptide loading; the catalyst-chaperone protein HLA-DM accelerates the rate of CLIP release and peptide exchange in MIIC. The transport of loaded MHC class II molecules is thought to involve the transformation of MIIC into tubular structures directed toward the site of T cell interaction at the plasma membrane.74
TLRs regulate phagosome maturation,75 enhance lysosomal acidification,73 and increase antigen uptake transiently.76 MHC class II surface expression and turnover rates are regulated by cytoplasmic domain ubiquitination in DCs.77 This explains the expression of low levels of MHC class II molecules by immature DCs and their increase in half-life after maturation, sustaining antigen presentation after migration into secondary lymphoid organs.
RA is characterized by autoantibodies against citrullinated proteins. A possible mechanism of generation of the immune response against citrullinated proteins has been recently described in mice.78 It has been shown that two distinct conformers of MHC-peptide complexes are known for the same peptide; these are referred to as type A for stable conformation and type B for less stable conformation. Peptides generated from intact protein are loaded onto MHC class II complex in the late endosome/lysosome. In this compartment, HLA-DM catalyzes peptide loading and acts as a conformational catalyst, hence only more stable type A conformers are loaded.78 In comparison, the same peptide may be loaded exogenously at the cell surface and in the early endosome in exchange for poorly binding peptides occupying MHC class II molecules in the absence of HLA-DM. These mechanisms of loading give rise to both stable type A and flexible type B conformers.78 Hence only type A T cells are primed with native protein, and exogenous peptide gives rise to priming of both type A and B T cells. In the thymus, T cells recognizing the type A conformation for a peptide are deleted, whereas type B T cells escape thymic negative selection.78 Thus self-reactive type B T cells are present in the naïve peripheral repertoire and have potential for autoimmune activation. This was recently shown in nonautoimmune prone mice after immunization with unmodified hen egg lysozyme (HEL); T cells specifically reactive to citrullinated epitopes of HEL were present among the responding repertoire to immunization.79 The citrullinated HEL epitopes were processed and loaded onto MHC class II complexes and were presented to T cells by DCs.79 Hence naturally occurring self-reactive T cells, which have evaded thymic negative selection, may recognize citrullinated peptides loaded as flexible type B conformers onto MHC class II molecules and subsequently provide help for B cell–mediated antibody responses against citrullinated proteins.
Lipid Presentation
CD1 molecules present lipid antigen to T cells. The CD1 family of MHC I-like glycoproteins includes CD1a-d on the surfaces of DCs and CD1e, which remains in the endoplasmic reticulum. CD1a-c–restricted T cells express CD4 or CD8 or lack both CD4 and CD8 (double negative). By contrast, most CD1d-restricted T cells express a semi-invariant T cell receptor (TCR) and markers of NK cells and are identified as invariant NK T (iNKT) cells.80
Similar to MHC I and MHC II molecules, CD1 molecules assemble in the ER, where they noncovalently bind the chaperones calnexin, calreticulin, ERp57, and β2 microglobulin.81 Lipid antigens are generated by lysosomal hydrolases of internalized phagosomes, membrane-bound lipids in clathrin-coated vesicles, or internalized apoptotic bodies or exosomes and are loaded onto CD1 molecules.
T Cell Activation
T cell activation is dependent on the intensity and length of DC–T cell interactions, mediated by the immunologic synapse (IS), which is a region of spatially and temporally organized motifs of membrane proteins and cytosolic molecules formed at the interface between a T cell and a DC.82 Stable long-lasting contacts of APCs with T cells are dependent on co-stimulatory molecules and integrin molecules ICAM-1 and ICAM-3. These integrins form a ring in the outer part of the IS, in a region adjacent to the corresponding area containing lymphocyte function associated antigen-1 (LFA-1).83 It is hypothesized that the stability of the IS confers temporal inhibition of DC apoptosis, which would enhance activation of clonal T cells in the lymph node.82
CD4+ T helper (Th) cells play a vital role in the induction of humoral and cellular immunity, providing signals for the activation of B cells and CD8+ T cells. Controlled by the activated DCs, CD4+ T cells can differentiate into Th1 cells, producing IFN-γ and IL-2; Th2 cells, producing IL-4, IL-5, and IL-13; follicular helper T cells (Tfh), expressing inducible T cell co-stimulator and producing IL-21; Th17 cells, producing IL-17; or regulatory T cells (Tregs), producing TGF-β or IL-10 (see Chapter 13).
DCs promote a Th1 phenotype by acting on both NK cells and naïve CD4+ T cells. Collaboration of IFN-γ and IL-12 induces and sustains Th1 differentiation. The Th2 response is promoted by induction of OX40 ligand (OX40L) on DCs, which is mediated by release of thymic stromal lymphopoietin (TSLP) by epithelial cells, mast cells, and basophils.84 Absence or depletion of IL-12 production by DCs also skews the T cell response to Th2. In turn, IL-10 secretion by Th2 cells can negatively regulate DC function.85 DCs make a deficient response to TSLP stimulation in Crohn’s disease, an inflammatory bowel disease associated with Th1 and Th17 immunity.86
Proinflammatory T cells expressing IL-17 were described in RA synovium as long ago as in 1998.87,88 Recently, Th17 cells expressing the RORγt transcription factor were described as a new subset of Th cells. Upon TLR stimulation, innate immune cells including DCs secrete IL-6 and IL-1β. The presence of IL-6 and TGF-β induces Th17 differentiation, subsequent production of IL-21, and expression of IL-23R.89 IL-1β is also a critical amplifier of Th17 differentiation, especially in humans. IL-1β and IL-6 also promote the reprogramming of FoxP3+ regulatory T cells to Th17.90 This scenario is relevant to the development of autoimmune disease, as Tregs are selected based on intermediate affinity for self-peptides in the thymus.91 IL-21 amplifies Th17 differentiation; however, IL-23—produced by DCs—is needed for maintenance of Th17 cells.89
Although Th1 cells were the major subset of T cells identified in RA synovium, identification of Th17 cells and the cytokines IL-17 and IL-23 within affected tissues and/or fluids has implicated both Th1 and Th17 cells in the pathogenesis of both RA and spondyloarthropathy.88,92 Synovial tissue DCs express IL-12p70 and IL-23p19, providing a mechanism by which Th1 and Th17 cell production is locally perpetuated.93
pDCs, potent producers of type I IFN, also present viral and tumor antigens to initiate both CD4+ and CD8+ T cell responses, thereby providing an important link between innate and adaptive immunity.94,95 Increased production of IFN-α may activate CD8+ T cells, which are increased in kidneys in lupus nephritis patients.96,97 In addition, pDCs can induce Treg generation by induction of indoleamine-2,3,-dioxygenase (IDO) (discussed later).









