Syndromes/disorders
OMIM
Gene
Acrofacial dysostosisa
263750
Alagille syndrome
118450
JAG1, NOTCH2
Anhalta
601344
Atelosteogenesis III
108721
FLNB
Campomelic dysplasia
211970
SOX9
Casamassima-Morton-Nancea
271520
Caudal regressiona
182940
Cerebro-facio-thoracic dysplasiaa
213980
CHARGE syndrome
214800
CHD7
‘Chromosomal’
Currarino
176450
HLXB9
Atelosteogenesis, type II (de la Chapelle syndrome)
256050
SLC26A2
DiGeorge/deletion 22q11.2/velocardiofacial syndrome
188400
Dysspondylochondromatosisa
Femoral hypoplasia-unusual faciesa
134780
Fibrodysplasia ossificans progressiva
135100
ACVR1
Fryns-Moermana
Goldenhar/OAV Spectruma
164210
Holmes-Schimkea
Incontinentia pigmenti
308310
IKBKG
Kabuki syndrome
147920
MLL2
McKusick-Kaufman syndrome
236700
MKKS
KBG syndrome
148050
ANKRD11
Klippel-Feila
148900
GDF6, GDF3, MEOX1
PAX1 b
Larsen syndrome
150250
FLNB
Lower mesodermal agenesisa
Maternal diabetes mellitusa
MURCS associationa
601076
Multiple pterygium syndrome
265000
CHRNG
OEIS syndromea
258040
Phavera
261575
RAPADILINO syndrome (RECQL4-related disorders)
266280
RECQL4
Robinow (ROR2-related disorders)
180700
ROR2
Rolland-Desbuquoisa
224400
Rokitansky sequencea
277000
WNT4 b
Silverman-Handmaker type of dyssegmental dysplasia (DDSH)
224410
HSPG2
Simpson-Golabi-Behmel syndrome
312870
GPC3
Sirenomeliaa
182940
Spondylocarpotarsal synostosis
272460
FLNB
Thakker-Donnaia
227255
Torielloa
Uriostea
VATER/VACTERLa
192350
Verloove-Vanhoricka
215850
Wildervancka
314600
Zimmera
301090
Although CS is frequently associated with SDV this is not always so and CS may occur in the absence of segmentation anomalies, though abnormalities of vertebral formation may be present. In cases of this kind a diagnosis of one of the skeletal dysplasias should be considered, though a precise radiological diagnosis may require follow-up skeletal surveys as the child grows. A clinical genetics opinion with a view to genetic testing may be very helpful and examples include: congenital contractural arachnodactyly (aka Beals syndrome), which is autosomal dominant and due to mutations in FBN2; chondrodysplasia punctata, Conradi-Hünermann type (aka Happle syndrome), which is X-linked and due to mutations in the EBP gene; diastrophic dysplasia, which is autosomal recessive and due to mutations in the sulphate transporter gene SLC26A2 (aka DTDST); and spondylometaphyseal dysplasia, Kozlowski type, which is autosomal dominant and due to mutations in TRPV4.
Spondylocostal Dysostosis, Somitogenesis, and the Notch Signaling Pathway
The main progress in understanding the genetic basis of SDV has come through the study of somitogenesis in animal models , mainly mouse but also chick. Animals with specific gene knockouts are generated and multiple gene expression assays undertaken to help elucidate the developmental pathways. Somitogenesis is the sequential process whereby paired blocks of paraxial mesoderm are patterned and laid down on either side of the midline from the presomitic mesoderm (PSM) to form somites, a process that takes place between days 20–32 of human embryonic development, proceeding in a rostro-caudal direction. In mouse, a pair of somites is formed every 1–3 h, whilst in humans the process is estimated to take 6–12 h based on cell culture models and analysis of staged anatomical collections [8, 9]. Somites ultimately give rise to four substructures—sclerotome , which forms the axial skeleton and ribs; dermotome, which forms the dermis; myotome, which forms the axial musculature and syndetome, which forms the tendons [10, 11]. Somitogenesis begins shortly after gastrulation and continues until the pre-programmed number of somite blocks is formed. In man 31 blocks of paired tissue are formed but the number is species-specific. The establishment of somite boundaries takes place as a result of very finely tuned molecular processes determined by activation and negative feedback interactions between components of the Notch, Wnt and FGF signaling pathways [12, 13] (Fig. 7.1). In the rostral third of the PSM formation of segmental boundaries is subject to levels of the factor FGF8, which is produced in the caudal region of the embryo [14] and which probably maintains cells in an immature state until levels fall below a threshold, allowing boundary formation. Somites already harbour specification toward their eventual vertebral identity, a process regulated by the Hox family of transcription factors [15], which also display oscillatory expression in the mouse during somitogenesis [16].
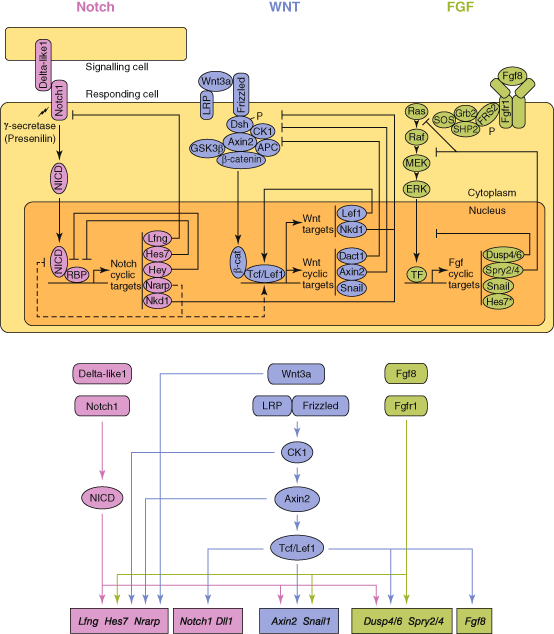
Fig. 7.1
The putative relationships between the Notch, Wnt and FGF pathways in somitogenesis. (Adapted from Gibb et al. [13] ©Elsevier)
The Wnt signalling pathway also displays oscillatory expression in a different temporal phase from Notch pathway genes, and plays a key role in the segmentation clock [17–19]. The mediators of the determination front and the segmentation clock (Notch, FGF, Wnt) are required for forming the somite boundary and specify rostro-caudal patterning of presumptive somites, for which Mesp2 is crucial [20]. Mesp2 is expressed caudal to the somite which is forming and this domain is set where Notch signalling is active, FGF signalling is absent, and the transcription factor Tbx6 is expressed. Precise periodicity in the establishment of somite blocks is mediated by several so-called ‘cycling’, or ‘oscillatory’, genes, two of which, LFNG and HES7, are implicated in human SCD.
Somites themselves, having formed, are subsequently partitioned into rostral and caudal compartments, with vertebrae formed from the caudal compartment of one somite and the adjacent rostral compartment of the next, a phenomenon that is known as ‘resegmentation’ [21–24]. An understanding of the molecular biology of somitogenesis in animal models, in combination with finding patients and families with specific forms, or patterns, of segmentation anomalies, has led to the most definitive progress in understanding the causes of rare mendelian forms of SDV. Ongoing research is identifying more cycling genes and pathways involved in the regulation of somitogenesis .
Varied Use of Clinical Terminology
Spondylocostal Dysostosis
In clinical practice the use of terms for vertebral segmentation abnormalities has been inconsistent and confusing . ‘Spondylocostal dysostosis’ (SCD) continues to be applied to a wide variety of radiological phenotypes where abnormal segmentation is evident together with rib involvement. For this review I use our preferred definition as given in Table 7.2. This restricts use of the term to generalised SDV , which defines the mendelian forms of SCD thus far identified, as summarised in Table 7.3. This is usually a short trunk, short stature condition with multiple/generalised SDV accompanied by rib fusions and/or mal-alignment. A mild, non-progressive kyphoscoliosis is present, usually without additional organ abnormalities. Five Notch signalling pathway genes are now linked to this group, four demonstrating autosomal recessive (AR) inheritance and one autosomal dominant (AD), as described below.
Table 7.2
Proposed definitions for the terms spondylocostal dysostosis (SCD) and spondylothoracic dysostosis (STD) (ICVAS)
Features | Spondylocostal Dysostosis (SCD) | Spondylothoracic Dysostosis (STD) |
|---|---|---|
General | No major asymmetry to chest shape | Chest shape symmetrical, with ribs fanning out in a ‘crab-like’ appearance |
Mild, non-progressive scoliosis | Mild, non-progressive scoliosis, or no scoliosis | |
Multiple SDV (MSDV) ≥ 10 contiguous segments | Generalised SDV (GSDV) | |
Absence of a bar | Regularly aligned ribs, fused posteriorly at the costovertebral origins, but no points of intercostal fusion | |
Mal-aligned ribs with intercostal points of fusion | ||
Specific, descriptive | ‘Pebble beach’ appearance of vertebrae in early childhood radiographs (Fig. 7.3) | ‘Tramline’ appearance of prominent vertebral pedicles in early childhood radiographs, not seen in SCD (Fig. 7.6) |
‘Sickle cell’ appearance of vertebrae on transverse imaging [49] |
Table 7.3
Genes causing generalised SDV, i.e. ‘spondylocostal dysostosis’ according to the definition proposed in Table 7.2
SCD | Gene symbol | Chromosomal locus | Protein name |
|---|---|---|---|
SCD type 1 | DLL3 | 19q13 | Delta-like protein 3 |
SCD type 2 and STD | MESP2 | 15q26.1 | Mesoderm posterior protein 2 |
SCD type 3 | LFNG | 7p22 | Beta-1,3-N-acetylglucosaminyltransferase lunatic fringe |
SCD type 4 | HES7 | 17p13.2 | Transcription factor HES-7 |
SCD type 5 | TBX6 | 16p11.2 | T-box6 protein |
A number of attempts have been made to classify SDV. The scheme proposed by Mortier et al. [25] combines phenotype and inheritance pattern (Table 7.4). The scheme proposed by Takikawa et al. [26] allows a very broad definition of SCD (Table 7.5), and both these schemes identify Jarcho-Levin Syndrome (JLS) with a ‘crab-like’ chest. McMaster and Singh’s [27] surgical approach to classification (1999) distinguishes between formation and segmentation errors (Table 7.6). As with McMaster’s scheme, Aburakawa’s [28] classification scheme for vertebral abnormalities (1996), which includes vertebral morphology (Table 7.7) , does not attempt to identify phenotypic patterns of malformation based on assessment of the spine as a whole. The use of a limited number of terms in these classification schemes neither reflects the great diversity of radiological SDV phenotypes seen in clinical practice nor incorporates knowledge from molecular genetics . Furthermore, the diversity of SDV is not captured within the classification of osteochondrodysplasias [29, 30]. A new scheme for classification and reporting from the International Consortium for Vertebral Anomalies and Scoliosis (ICVAS) is described later .
Nomenclature | Definition |
|---|---|
Jarcho-Levin syndrome | Autosomal recessive |
Symmetrical crab-like chest, lethal | |
Spondylothoracic dysostosis | Autosomal recessive |
Intrafamilial variability, severe/lethal | |
Associated anomalies uncommon | |
Spondylocostal dysostosis | Autosomal dominant |
Benign | |
Heterogeneous group | Sporadic |
Associated anomalies common |
Nomenclature | Definition |
|---|---|
Jarcho-Levin syndrome | Symmetrical crab-like chest |
Spondylocostal dysostosis | ≥ 2 vertebral anomalies associated with rib anomalies (fusion and/or absence) |
Table 7.6
Classification (surgical/anatomical) of vertebral segmentation abnormalities causing congenital kyphosis/kyphoscoliosis, according to McMaster and Singh [27]
Type | Anatomical deformity | Anomalies |
|---|---|---|
I | Anterior failure of vertebral body formation | Posterolateral quadrant vertebrae |
Single vertebra | ||
Two adjacent vertebrae | ||
Posterior hemivertebrae | ||
Single vertebra | ||
Two adjacent vertebrae | ||
Butterfly (sagittal cleft) vertebrae | ||
Anterior or anterolateral wedged vertebrae | ||
Single vertebra | ||
Two adjacent vertebrae | ||
II | Anterior failure of vertebral body segmentation | Anterior unsegmented bar |
Anterolateral unsegmented bar | ||
III | Mixed | Anterolateral unsegmented bar contralateral posterolateral quadrant vertebrae |
IV | Unclassifiable |
Failure of formation |
Type I |
A. Double pedicle |
B. Semi segmented |
C. Incarcerated |
Type II |
D. Non incarcerated, no lateral shift |
E. Non incarcerated, plus lateral shift |
Type III |
F. Multiple |
Type IV |
G. Wedge |
H. Butterfly |
Failure of segmentation |
I. Unilateral Bar |
J. Complete block |
K. Wedge (plus narrow disc) |
Mixed |
L. Unilateral bar plus hemivertebra |
M. Unclassifiable |
Klippel-Feil Syndrome
The term Klippel-Feil anomaly or syndromes (KFS) has a more specific application, even though the phenotypes within the general category are diverse. KFS refers to vertebral fusion or segmentation errors involving the cervical region and has been the subject of several classifications (Table 7.8) [31, 32]. Clarke et al. [33] (Table 7.9) proposed a further, detailed classification combining modes of inheritance. To these clinical classifications must now be added a classification based on the recently discovered gene associations with rare forms of KFS [34–36] (Table 7.10). The Pax1 gene has been shown to be active during sclerotome formation and differentiation and mutations were identified in the mouse undulated, suggesting that sclerotome condensation is a Pax1-dependent process [37]. Two studies on patient cohorts with KFS were subsequently undertaken [6, 38] but despite some gene variants being identified in a small number the same variants were either detected in an asymptomatic parent or did not occur in a conserved region of the gene. Overall, the role of PAX1 in KFS remains to be elucidated .
Type | Site | Anomaly |
|---|---|---|
I | Cervical and upper thoracic | Massive fusion with synostosis |
II | Cervical | One or two interspaces only, hemivertebrae, occipito-atlantoid fusion |
III | Cervical and lower thoracic or lumbar | Fusion |
Table 7.9
Classification of Klippel-Feil anomaly according to Clarke et al. [33]. (Adapted from original publication)
Class | Vertebral fusions | Inheritance | Possible anomalies |
|---|---|---|---|
KF1 | Only class with C1 fusions C1 fusion not dominant Variable expression of other fusions | Recessive | Very short neck; heart; urogenital; craniofacial; hearing; limb; digital; ocular defects Variable expression |
KF2 | C2-3 fusion dominant C2-3 most rostral fusion Cervical, thoracic and lumbar fusion variable within a family | Dominant | Craniofacial; hearing; otolaryngeal; skeletal and limb defects Variable expression |
KF3 | Isolated cervical fusions Variable position Any cervical fusion except C1 | Recessive or reduced penetrance | Craniofacial Facial dysmorphology Variable expression |
KF4 | Fusion of cervical vertebrae, data limited | Possible X-linked Predominantly females | Hearing and ocular anomalies—abducens palsy with retraction bulbi aka Wildervanck syndrome |
KFS | Gene symbol | Chromosomal locus | Encodes | Inheritance |
|---|---|---|---|---|
KFS1 | GDF6 [aka cartilage-derived morphogenetic protein 2 (CDMP2)] | 8q22.1 | A member of the bone morphogenetic protein family | AD |
KFS2 | MEOX1 | 17q21.31 | Homeodomain-containing protein | AR |
KFS3 | GDF3 | 12p13.1 | A member of the bone morphogenetic protein family
Related posts: The Genetic Architecture of Idiopathic Scoliosis The Genetic Architecture of Idiopathic Scoliosis
 Molecular Genetics of Congenital Multiple Large Joint Dislocation Molecular Genetics of Congenital Multiple Large Joint Dislocation
 Overview of Next Generation, High-Throughput Molecular Genetic Methods Overview of Next Generation, High-Throughput Molecular Genetic Methods
 Neurofibromin in Skeletal Development Neurofibromin in Skeletal Development
 DMP-1 in Postnatal Bone Development DMP-1 in Postnatal Bone Development
 Genetic and Environmental Interaction in Malformation of the Vertebral Column Genetic and Environmental Interaction in Malformation of the Vertebral Column
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|