25 Cell Recruitment and Angiogenesis
A number of cell adhesion molecules are involved in leukocyte extravasation.
Angiogenesis, the formation of new vessels, is involved in inflammation and tumor progression.
Inflammatory leukocytes, endothelial cells (ECs), synovial fibroblasts, and soluble mediators and cell adhesion molecules (CAMs) are involved in cell trafficking into inflammatory sites in diseases such as rheumatoid arthritis (RA)1–5 (Figure 25-1). In arthritis, leukocyte ingress into the synovium occurs by leukocyte adhesion to ECs and then by transendothelial migration.3–5 The chemotaxis of inflammatory cells is mainly regulated by chemotactic mediators termed chemokines.6–10 The formation of new capillaries from preexisting vessels, termed angiogenesis, is a key event underlying synovial inflammation, which perpetuates the recruitment of leukocytes into the synovium.7,11–15 On the other hand, new vessel formation from endothelial progenitor cells (EPCs), termed vasculogenesis, is impaired in inflammatory arthritides.15–19 Several CAMs that interact with each other, as well as with soluble inflammatory mediators such as cytokines and chemokines, are involved in synovial leukocyte recruitment and angiogenesis.3,7,12,20
Endothelial Pathophysiology In Inflammation
Endothelial cells are active players in inflammation. The vascular endothelium undergoes vasodilation and increased permeability (leakage) during synovitis.21,22 Increased endothelial permeability results from several mechanisms including endothelial contraction and retraction, leukocyte- or antiendothelial antibody (AECA)-mediated vascular injury and regeneration.21–23 The endothelium secretes several inflammatory mediators resulting in vasodilatation and leakage including prostacyclin (PGI2), nitric oxide (NO), platelet-activating factor (PAF), and others.21,23 In turn, endothelial cells respond to histamine, serotonin, complement factors (C3a, C5a), bradykinin, leukotrienes, PAF, and AECAs that are released in inflammatory sites.21–23 Cytoskeletal reorganization leading to endothelial retraction may be regulated by proinflammatory cytokines such as interleukin (IL)-1, tumor necrosis factor (TNF), or interferon-γ (IFN-γ).21–23 Among other soluble mediators, AECAs that have been detected in several inflammatory rheumatic conditions have been correlated with clinical activity and vascular damage.24,25
High-endothelial venules (HEVs) are usually detected in lymphoid tissues, and they are major sites of leukocyte extravasation during the homing process.26,27 Such HEVs have been described at least in some synovial tissues with lymphoid neogenesis.28,29
Intercellular Adhesion Molecules
Process of Leukocyte Extravasation in Inflammation
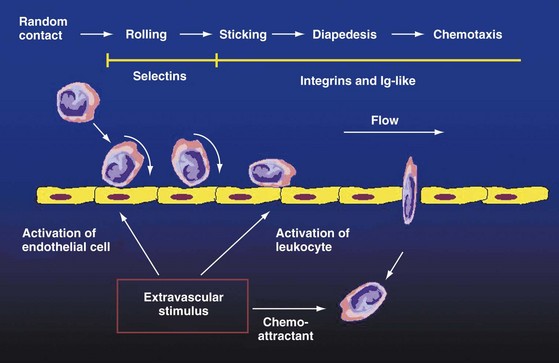
Adhesion of peripheral blood leukocytes to endothelium leads to the process of leukocyte transendothelial migration into inflammatory sites such as the arthritic synovium.2–4 High endothelial venules (HEVs) primarily found in lymphoid organs are also present at sites of lymphoid neogenesis in the synovium.3,29,30 Lymphocytes recirculate through HEVs during homing, and thus inflammatory leukocyte recruitment may be considered as “pathologic homing”3,4,29 (see Figure 25-1).
During leukocyte adhesion and transendothelial migration, an early, weak adhesion termed rolling occurs first. This step involves selectins and their ligands and leads to leukocyte activation. Activation-dependent, firm adhesion involves mostly integrin-dependent interactions, as well as the secretion of numerous chemokines. Chemokines preferentially attract endothelium-bound leukocytes2,20,31 (see Figure 25-1).
Adhesion Receptors and Ligands
CAMs have been classified into integrin, selectin, immunoglobulin, and cadherin superfamilies4,32 (Table 25-1). E-, P-, and L-selectin contain a lectin-like extracellular N-terminal domain, an epidermal growth factor (EGF)-like motif, and two to nine moieties related to complement regulatory proteins.33,34 E- and P-selectin are expressed by ECs, whereas L-selectin is mostly expressed by leukocytes.34 During leukocyte transendothelial migration, selectins mediate the initial tethering and rolling of leukocytes.20,34,35 E-selectin is a marker for cytokine-induced EC activation.34 E-selectin ligand-1 (ESL-1) and P-selectin ligand-1 (PSGL-1) contain sialylated glycan motifs such as sialyl Lewis-X (sLex).34 P-selectin is constitutively present on the membrane of EC Weibel-Palade bodies.34 P-selectin is involved in the early phases of leukocyte-EC adhesion.35 L-selectin serves as a lymphocyte homing receptor, where it mediates the physiologic recirculation of naïve lymphocytes through specialized HEV.32,34 However, L-selectin has also been implicated in inflammatory leukocyte recruitment.3,34 All three selectins are expressed in the arthritic synovium3,4,36 (see Table 25-1).
Table 25-1 Relevant Members of Adhesion Molecule Superfamilies*
| Adhesion Receptors | Ligands |
|---|---|
| Selectins | |
| L-selectin (CD62L, LAM-1) | Sialylated carbohydrates, GlyCAM-1 |
| E-selectin (CD62E, ELAM-1) | Sialyl-Lewis-X |
| P-selectin (CD62P, PADGEM) | Sialyl-Lewis-X, other carbohydrates |
| Integrins | |
| α1β1 (VLA-1) | Laminin, collagen |
| α2β1 (VLA-2) | Laminin, collagen |
| α3β1 (VLA-3) | Laminin, collagen, fibronectin |
| α4β1 (VLA-4) | Fibronectin, VCAM-1 |
| α5β1 (VLA-5) | Fibronectin |
| α6β1 (VLA-6) | Laminin |
| αLβ2 (LFA-1, CD11a/CD18) | ICAM-1, ICAM-2, ICAM-3, JAM-A |
| αMβ2 (Mac-1, CD11b/CD18) | ICAM-2, iC3b |
| αXβ2 (CD11c/CD18) | iC3b, fibrinogen |
| αEβ7 | E-cadherin |
| α4β7 | Fibronectin, VCAM-1, MadCAM-1 |
| Immunoglobulin Superfamily | |
| ICAM-1 (CD54) | LFA-1, Mac-1 |
| ICAM-2 | LFA-1 |
| ICAM-3 | LFA-1 |
| VCAM-1 | α4β1, α4β7 |
| MadCAM-1 | α4β7, L-selectin |
| CD2 | LFA-3 |
| PECAM-1 (CD31) | PECAM-1, αVβ3 |
| Cadherins | |
| E-cadherin (cadherin-1) | E-cadherin |
| N-cadherin (cadherin-2) | N-cadherin |
| Cadherin-11 | Cadherin-11 |
Modified from Agarwal SK, Brenner MB: Role of adhesion molecules in synovial inflammation. Curr Opin Rheumatol 18(3):268–276, 2006.
Integrins are αβ heterodimers. Each of the common β chains is associated with one or more α subunits.3,32 Cell adhesion to the extracellular matrix (ECM) is mostly mediated by β1 and β3, whereas intercellular adhesion is assisted through β1 and β2 integrins.2,32 β1 and β3 integrins are expressed on ECs, whereas β2 integrins are leukocyte CAMs.32 Integrin-mediated adhesion and migration have been associated with arthritis.3,4,37 The α1-α6, αV, αL, αM, αX, and β1-β7 integrin subunits have all been detected in the inflamed synovium.3,4,37
The immunoglobulin superfamily of CAMs is a group of transmembrane glycoproteins containing one or more immunoglobulin-like motifs of 60 to 100 amino acids.32 Vascular cell adhesion molecule-1 (VCAM-1) is constitutively expressed on ECs; however, its expression is upregulated by proinflammatory cytokines.38 There is abundant VCAM-1 expression in the inflamed synovium.3,37,39 ICAM-1, the counterreceptor for the β2 integrins LFA-1 (αLβ2), Mac-1 (αMβ2), and αXβ2, is expressed on both ECs and leukocytes.3,32,40 ICAM-1 is highly expressed on ECs in inflammatory sites such as in the RA synovium.37,40 Among other ICAMs, ICAM-2 is constitutively expressed on ECs and may not be an activation marker.40 ICAM-3 is a leukocyte CAM; however, it is also present on some RA synovial ECs.40 All three ICAMs bind to β2 integrins.3,40 Other members of this superfamily include CD2 and LFA-3. CD2 binds to LFA-3, and both CAMs exert abundant expression in the arthritic synovium.3,41 Platelet-endothelial adhesion molecule 1 (PECAM-1 or CD31) mediates homotypic adhesion by binding to another PECAM-1 molecule, as well as heterotypic adhesion by recognizing the αVβ3 integrin.3,32,42 PECAM-1 is a marker of endothelial activation, and it is expressed in the arthritic synovium.42,43
Some other CAMs involved in leukocyte-EC adhesion underlying inflammation and associated with arthritis include CD44, vascular adhesion proteins (VAP-1 and VAP-2), endoglin, E- and N-cadherin, cadherin-11, and junctional adhesion molecules (JAMs).2–4,36,41,44–48 CD44 is a receptor for hyaluronate32 and is expressed on activated ECs in inflammatory sites.36,41,49,50 VAP-1 has originally been isolated from synovial ECs. The expression of VAP-1 is increased in RA.46,51 Endoglin (CD105) is a receptor for transforming growth factor (TGF)-β1. Endoglin is involved in EC adhesion and is expressed in synovitis.52,53 Cadherins are primarily involved in embryogenesis; however, synovial fibroblast cadherin-11 has been implicated in arthritis as well4,54,55 (see Table 25-1).
Chemokines and Chemokine Receptors
Chemokines are small proteins that exert chemotactic activity toward leukocytes.9,12,56,57 There are four known chemokine supergene families based on the location of cysteine (C) residues within the chemokine structure. These families are designated as CXC, CC, CX3C, and C chemokines; their respective chemokine receptor groups are CXCR, CCR, CX3CR, and CR; and the current designation of chemokine members are CXCL, CCL, CX3CR, and XCL.9,56 More than 50 chemokines and 19 chemokine receptors have been identified9,56 (Table 25-2).
Table 25-2 Chemokine Receptors with Their Most Relevant Ligands*
| Chemokine Receptor | Chemokine Ligand |
|---|---|
| CXC Chemokine Receptors | |
| CXCR1 | CXCL8 (IL-8) |
| CXCR2 | CXCL8, CXCL5 (ENA-78), CXCL1 (GROα), CXCL7 (CTAP-III) |
| CXCR3 | CXCL10 (IP-10), CXCL4 (PF4), CXCL9 (Míg), CXCL11 (ITAC) |
| CXCR4 | CXCL12 (SDF-1) |
| CXCR5 | CXCL13 (BCA-1) |
| CXCR6 | CXCL16 |
| CXCR7 | CXCL12, CXCL11 |
| C-C Chemokine Receptors | |
| CCR1 | CCL3 (MIP-1α), CCL5 (RANTES), CCL7 (MCP-3), CCL23 (MPIF-1) |
| CCR2 | CCL2 (MCP-1), CCL7 |
| CCR3 | CCL5, CCL8 (MCP-2) |
| CCR4 | CCL17 (TARC), CCL22 (MDC) |
| CCR5 | CCL3, CCL4 (MIP-1β), CCL5 |
| CCR6 | CCL20 (MIP-3α) |
| CCR7 | CCL19 (MIP-3β), CCL21 (SLC) |
| CCR8 | CCL1 (I-309) |
| CCR9 | CCL25 (TECK) |
| CCR10 | CCL27 (CTACK), CCL28 (MEC) |
| C Chemokine Receptors | |
| XCR1 | XCL1 (Lymphotactin) |
| C-X3-C Chemokine Receptors | |
| CX3CR1 | CX3CL1 (Fractalkine) |
Modified from Koch AE: Chemokines and their receptors in rheumatoid arthritis: future targets? Arthritis Rheum 52(3):710–721, 2005.
Chemokine Superfamilies
Most CXC chemokines chemoattract neutrophils. Many genes coding these chemokines are clustered on chromosome 4q12-13.57 In contrast, the genes of some CXC chemokines such as CXCL4 (platelet factor 4; PF4) and CXCL10 (IFN-γ-inducible 10-kD protein; IP-10) are located on different chromosomes, and these chemokines recruit lymphocytes and monocytes.56,57
CC chemokines stimulate monocyte chemotaxis, but some members of this subclass may also recruit lymphocytes. The genes of monocyte-chemoattracting CC chemokines have been clustered to chromosome 17q11.2. In contrast, genes of CC chemokines recruiting lymphocytes are generally located elsewhere.9,56,57
The CX3C chemokine family has only one member, CX3CL1 (fractalkine).12,56,58–60 This chemokine is chemotactic for mononuclear cells, but it also serves as an adhesion molecule.59,60
The C family contains two members: XCL1 (lymphotactin) and XCL2 (single C motif 1β; SCM-1β). Lymphotactin is primarily involved in the migration of T lymphocyte subsets to inflammatory sites.61,62
Chemokine Receptors
The chemokines described earlier mediate their effects via 7-transmembrane domain receptors expressed on the target cells.56,57 Although some receptors (e.g., CXCR2, CCR1, CCR3) have multiple chemokine ligands, others (e.g., CXCR6, CCR8, CCR9) are specific receptors for one single ligand.9,57 Again, there may be a relationship between some chemokine receptors and the functions of their ligand(s). For example, single-ligand receptors such as CCR8 or CCR9 bind to chemokine ligands mostly exerting homeostatic functions (see later). In contrast, CXCR2, a receptor recognizing multiple CXC chemokines, plays a crucial role in inflammation and angiogenesis.9,63 Chemokine receptors have also been associated with various types of autoimmune inflammation. For example, RA, a mostly Th0-Th1 type disease, is associated with CXCR3 and CCR5, whereas asthma, a known Th2 type disease, is rather associated with CCR3, CCR4, and CCR8 tissue expression.12,64,65
Inflammatory and Homeostatic Chemokines: Is It a Justified Classification?
Chemokines have recently been functionally classified into these subgroups.10,56 As many functions of these chemokines overlap, this classification may not be really justified. Numerous CXC, CC, and CX3C chemokines implicated in the pathogenesis of arthritis are termed inflammatory chemokines. These chemokines recruit mostly effector cells including monocytes, neutrophils, and T cells into tissues.9,10,56,66 As included in Table 25-2, there is a great body of evidence suggesting the role in RA of CXCL1 (growth-regulated oncogene α; GROα); CXCL4 (platelet factor 4; PF4); CXCL5 (epithelial-neutrophil activating protein 78; ENA-78); CXCL6 (granulocyte chemotactic protein 2; GCP-2); CXCL7 (connective tissue activating protein III; CTAP-III); CXCL8 (interleukin 8; IL-8); CXCL9 (monokine induced by interferon-γ; Mig); CXCL10 (interferon-γ-inducible 10-kD protein; IP-10); CXCL12 (stromal cell–derived factor 1; SDF-1); CXCL13 (B cell–activating chemokine 1; BCA-1); and CXCL16. Among CC chemokines, CCL2 (monocyte chemoattractant protein 1; MCP-1); CCL3 (macrophage inflammatory protein 1α; MIP-1α); CCL5 (Regulated upon Activation, Normal T cell Expressed and Secreted; RANTES); CCL19 (Epstein-Barr virus–induced gene 1 ligand chemokine; ELC); CCL20 (MIP-3α); and CCL21 (secondary lymphoid tissue chemokine; SLC) have been implicated in leukocyte recruitment underlying inflammatory synovitis. Finally, CX3CL1 (fractalkine) and XCL1 (lymphotactin) are also considered as inflammatory chemokines.7,9,56 Accordingly, CXCR1-CXCR6, CCR1-CCR6, and XCR1 and CX3CR1 are involved in the pathogenesis of RA.7,56,67
Homeostatic chemokines are constitutively produced in microenvironments of lymphoid or nonlymphoid tissues such as in the skin or mucosa. These chemokines promote lymphocyte homing into these tissues, a process associated with the physiologic function of the adaptive immune system. Lymphocyte recirculation is involved in antigen sampling and immune surveillance.10,68,69 Among CXC chemokines, CXCL12 (SDF-1), CXCL13 (BCA-1), and CXCL16, as well as their respective receptors, CXCR4, CXCR5, and CXCR6, exert such effects.10,68,69 Among CC chemokines, CCL17 (TARC), CCL19 (ELC), CCL21 (SLC), CCL22 (MDC), CCL25 (TECK), CCL27 (CTACK), CCL28 (MEC), as well as their receptors, CCR4, CCR7, CCR9, and CCR10, are involved in the homeostasis of lymphoid tissues.56,68,69 As described earlier, among others, CXCL12, CXCL13, CXCL16, CCL19, and CCL21 have also been implicated in arthritis-associated inflammatory cell recruitment and synovial lymphoid neogenesis.7,10,56,67–71 The synovium is, in many ways, similar to mucosa-associated lymphoid tissues (MALT), which may explain the dual role of some chemokines in physiologic lymphocyte homing and inflammation.10,68,69,71
Angiogenesis and Vasculogenesis in Inflammation
Angiogenesis and Vasculogenesis
Angiogenesis is the formation of new capillaries from preexisting blood vessels, whereas vasculogenesis is the outgrowth of vessels from endothelial progenitor cells (EPCs).7,13–15,72–77 Angiogenesis may increase the total endothelial surface and thus may enable leukocyte extravasation into inflammatory sites.14,15 The perpetuation of angiogenesis has been associated with inflammatory diseases such as RA or psoriasis and in malignancies.14,15,77,78 The outcome of such “angiogenic diseases” is dependent on the balance or imbalance between angiogenic mediators and angiostatic factors.77 Several cytokines, growth factors, chemokines, certain CAMs, and other mediators can modulate neovascularization in inflammation14,15,73,77 (Table 25-3; see Figure 25-1).
Table 25-3 Angiogenic and Angiostatic Factors in Rheumatoid Arthritis*
| Mediators | Inhibitors | |
|---|---|---|
| Chemokines | CXCL1, CXCL5, CXCL7, CXCL8, CXCL12, CCL2, CCL21, CCL23, CX3CL1 | CXCL4, CXCL9, CXCL10, CCL21 |
| Matrix molecules | Type I collagen, fibronectin, laminin, heparin, heparan sulfate | Thrombospondin, RGD sequence |
| Cell adhesion molecules | β1 and β3 integrins, E-selectin, P-selectin, CD34, VCAM-1, endoglin, PECAM-1, VE-cadherin, Ley/H, MUC18 | RGD sequence (integrin ligand) |
| Growth factors | VEGF, bFGF, aFGF, PDGF, EGF, IGF-I, HIF-1, TGF-β† | TGF-β† |
| Cytokines | TNF, IL-6†, IL-15, IL-18 | IL-4, IL-6†, IFN-α, IFN-γ |
| Proteases | MMPs, plasminogen activators | TIMPs, plasminogen activator inhibitors |
| Others | Angiogenin, substance P, prolactin | DMARDs, TNF blockers, angiostatin, endostatin |
* See text for abbreviations. See Table 25-2 for traditional chemokine designations.
† Mediators with both proangiogenic and antiangiogenic effects.
Angiogenic Factors
The hypoxia-vascular endothelial growth factor (VEGF)-angiopoietin system seems to be of outstanding importance in arthritis-associated angiogenesis.73,79,80 VEGF is induced by hypoxia and hypoxia-inducible factors 1 and 2 (HIF-1, HIF-2) in RA.11,73,80–85 Significant hypoxia is characteristic of RA joints.73,86 Recently, the stimulatory effect of hypoxia on the angiogenic drive of RA synovial fibroblasts has been demonstrated.84,87 Hypoxia-inducible HIF-1 and HIF-2 are strongly expressed in the RA synovium.81,84,85,87 However, hypoxia may also act via HIF-independent regulatory pathways including the peroxisome-proliferator-activated receptors (PPARs).88 The angiopoietin-1 (Ang1)/Tie2 complex interacts with VEGF during the stabilization of newly formed blood vessels.89 In contrast, Ang2, an antagonist of Ang1, inhibits vessel maturation.80,89 Ang1 and Tie2 have been detected in the RA synovium.90,91
Apart from VEGF, other growth factors including fibroblast growth factors (FGF-1 and FGF-2), transforming growth factor β (TGF-β), connective tissue growth factor (CTGF), and platelet-derived growth factor (PDGF) have been implicated in synovial angiogenesis.1,7,14 Recently, the role of placenta growth factor (PIGF) in RA and inflammatory angiogenesis has been postulated. PIGF, like VEGF, binds to the flt-1/VEGF-R1 receptor. There is abundant expression of PIGF in the synovial tissue of arthritic mice.92
Proinflammatory cytokines may exert direct angiogenic activity or may act indirectly via VEGF-dependent pathways.14,93 Primarily TNF, IL-1, IL-6, IL-15, IL-17, IL-18, oncostatin M, granulocyte (G-CSF), and granulocyte-macrophage colony-stimulating factors (GM-CSFs) have been implicated in synovial angiogenesis.14,93 Monocyte migration inhibitory factor (MIF) has been implicated in angiogenesis, as well as atherosclerosis, a major cause of death in RA patients.94,95
Among numerous other angiogenic mediators not mentioned earlier, serum amyloid A (SAA), an important acute-phase reactant, has also been implicated in arthritis and angiogenesis. Interaction of SAA with formyl peptide receptor-like 1 (FPRL1) induces endothelial cell proliferation, migration and angiogenesis, and synovitis.96,97 Further angiogenic factors include, without mentioning further details, endothelin 1 (ET-1), members of the cyclooxygenase-2 (COX-2)-prostaglandin E2 network, angiogenin, angiotropin, pleiotrophin, platelet-activating factor (PAF), substance P, erythropoietin, adenosine, histamine, prolactin, thrombin, and sphingosine-1-phosphate (S1P).1,14,15,98
Vasculogenesis in Inflammatory Conditions
EPCs are hematopoietic stem cells expressing, among other antigens, CD34, CD133, type 2 VEGF receptor (VEGFR-2 or Flk-1), and the CXCR4 chemokine receptor.15–17,99–101 During vasculogenesis, EPCs differentiate into mature endothelial cells.101 Vasculogenesis is involved in tissue development, vascular repair, atherosclerosis, and inflammation.15,17,99–102
Several groups have described defective vasculogenesis related to impaired EPC numbers and functions in RA and scleroderma.1,16,17,102–105 Impaired vasculogenesis has been associated with increased cardiovascular morbidity and mortality in these disease states.1,102,106 Effective control of inflammation using corticosteroids and anti-TNF agents may stimulate EPCs and thus may restore defective vasculogenesis.103,107 In addition, the induction of vasculogenesis may be beneficial for patients with cardiovascular disease106 and the stimulation of EPCs and vasculogenesis may also suppress premature atherosclerosis in RA.1
Interactions among Adhesion Receptors, Chemokines, and Angiogenesis: The “Real” Bermuda Triangle in the Regulation of Inflammatory Synovitis
Chemokines and Adhesion Receptors
The molecular mechanisms and signaling pathways of chemokine-induced CAM expression have been described. Briefly, an atypical protein kinase C, PKC-ξ, has been identified. Treatment of cells with chemokines induces PKC-ξ kinase activity through its interaction with PI3K. This leads to increased cell surface integrin expression via further signaling steps.1
Another example for chemokine-CAM interactions is the regulation of β3 integrin expression by CCL2 (MCP-1) through the Ets-1 transcription factor, and the ERK-1/2 cascade. This pathway is involved in both inflammation and angiogenesis.108 The CCL21-CCR7 interaction results in the stimulation of LFA-1- and ICAM-1-dependent adhesion.109 Stimulation of CXCR1- and CXCR2-dependent pathways in the antigen-induced arthritis (AgIA) model resulted in increased neutrophil adhesion to endothelium.110 Thus various chemokines and chemokine receptors are involved in driving leukocyte transendothelial migration.
Chemokines and Chemokine Receptors in Angiogenesis and Vasculogenesis
Numerous CXC and CC chemokines, fractalkine, and their receptors have been implicated in angiogenesis underlying RA.7,12,14,111 The angiogenic nature of most CXC chemokines has been associated with the glutamyl-leucyl-arginyl (ELR) amino acid motif within their structure.63 ELR-containing CXC chemokines that mediate angiogenesis include CXCL1 (GROα), CXCL5 (ENA-78), CXCL7 (CTAP-III), and CXCL8 (IL-8).12,15,63,111 In contrast, the ELR-lacking CXCL4 (PF4), CXCL9 (Míg), and CXCL10 (IP-10) inhibit angiogenesis.8,9,39 Interestingly, some authors suggest that the effect of VEGF on endothelial cells may be, in part, mediated by CXCL10.44 It seems that CXCL12 (SDF-1) may play a significant role in RA-associated angiogenesis despite the fact that this chemokine lacks the ELR motif.7,112,113 CXCL12 is also a major mediator of lymphoid neogenesis in the RA synovium.112,113 Hypoxia stimulates CXCL12 production by RA synovial fibroblasts.112 CXCL12 has even been implicated in vasculogenesis. Virtually all EPCs express CXCR4 and migrate in response to SDF-1/CXCL12.42,43
Much less evidence is available regarding the role of CC chemokines in angiogenesis. CCL2 (MCP-1) may induce endothelial cell chemotaxis in vitro and angiogenesis in vivo.8,47 As described earlier, CCL2-induced angiogenesis may occur via β3 integrins.108 CCL23 has been implicated in the migration of vascular endothelial cells and angiogenesis-associated matrix metalloproteinase (MMP) production.48 In contrast, CCL21 (SLC) may exert strong angiostatic and antitumor effects.49
CX3CL1 (fractalkine) is involved in both angiogenesis and atherosclerosis underlying inflammatory rheumatic diseases.8,37,38
Adhesion Receptors, Ligands, and Proteases in Angiogenesis and Vasculogenesis
Both CAM receptors and their extracellular matrix (ECM) macromolecule ligands mediate adhesive interactions during inflammatory neovascularization.4,14,114 Among ECM components, type I and other minor collagens, fibronectin, heparin, laminin, tenascin, vitronectin, and fibrinogen promote angiogenesis.13,114 Vasculogenesis is stimulated by the laminin matrix Matrigel.115 Thrombospondin-1 (TSP-1) is an angiostatic ECM component naturally produced within the RA synovium.14,116,117
Among endothelial CAMs, soluble E-selectin; soluble P-selectin; the L-selectin ligand CD34; soluble VCAM-1; some endothelial β1, β3, and β5 integrins; PECAM-1 (CD31); endoglin (CD105); and some cadherins have been implicated in angiogenesis.13,14,118–121 The αVβ3 integrin and the ITGAV gene play critical roles in inflammatory angiogenesis. The integrin has become a major target for specific therapy.108,120–122 Other angiogenic factors such as chemokines may act via integrin-dependent pathways.108 Recently, the role of the ITGAV allele in angiogenesis has been further analyzed in four Caucasian sample sets. The genetic association could not be confirmed in New Zealand and Oxford (UK) sample sets, suggesting that the link between ITGAV gene polymorphism and RA may be limited.120 Focal adhesion kinases (FAKs) are involved in αVβ3 integrin signaling underlying synovial inflammation and angiogenesis.123 Among glycoconjugates with adhesive properties, blood group antigens Lewis-y and Lewis-H promote neovascularization.124 JAMs (JAM-A, JAM-B, and JAM-C) have also been implicated in the adhesive processes underlying RA, as well as in synovial angiogenesis.45,125 Vasculogenesis also involves numerous integrins including αVβ3 and E-selectin.1,99,126–128
MMPs promote angiogenesis by synovial matrix degradation.14,87,129,130 The role of hypoxia in MMP production is described earlier.87 Some ADAM and ADAMTS proteases have also been implicated in inflammatory neovascularization.131–133
Targeting Cell Adhesion, Chemokines, and Angiogenesis: Possible Therapeutic Approaches in Inflammatory Arthritides
Leukocyte recruitment inhibition may be a result of nonspecific anti-inflammatory therapeutic strategies. Numerous traditional and biologic disease-modifying drugs (DMARDs) and immunosuppressive agents may, in addition to other effects, suppress leukocyte recruitment, chemokine production, and angiogenesis. Inhibition of cell adhesion and migration, angiogenesis, chemokines, and chemokine receptors using specific antibodies or purified ligands has provided an important perspective on the molecular pathogenesis of RA. In addition, some of these strategies may be included in the future therapy of arthritis.*
Inhibition of Cell Adhesion Receptors and Leukocyte-Endothelial Adhesion
Traditional DMARDs including sulfasalazine, methotrexate (MTX), and leflunomide suppressed serum and synovial fluid soluble ICAM-1, VCAM-1, and E-selectin levels in both early and established RA, as well as juvenile idiopathic arthritis (JIA).136–139 MTX and leflunomide also decrease synovial tissue CAM expression in RA.140,141 Statins, currently used for the treatment of dyslipidemia, may also modify endothelial function and CAM expression.142
Infliximab therapy reduced the serum levels of soluble ICAM-1, ICAM-3, VCAM-1, and E- and P-selectin in RA and JIA.143–146 Adalimumab therapy resulted in the attenuation of neutrophil chemotaxis in RA.147 Abatacept treatment also reduced soluble ICAM-1 and E-selectin levels in RA.148 Tocilizumab also acts, in part, by inhibiting leukocyte recruitment.149
Regarding specific anti-CAM targeting in humans, first an antihuman ICAM-1 antibody (enlimomab) was used to treat refractory RA. Many patients reported improvement in their status; however, repeated administration of this antibody resulted in diminished efficacy and frequent adverse events. Therefore further development of enlimomab in RA was terminated.150,151 Two anti-integrin strategies, the anti-LFA-1 antibody efalizumab and the LFA-3-Ig fusion protein alefacept, have been registered for the treatment of psoriasis.152,153 Alefacept yielded to a moderate effect in psoriatic arthritis.154,155 Efalizumab was withdrawn from the market in 2009 due to severe side effects. Other anti-LFA-1 antibodies have still been in preclinical arthritis studies.156 Natalizumab (anti-α4 integrin) has been tried in multiple sclerosis and Crohn’s disease,4,69 and a monoclonal antibody to the α4β7 integrin was administered to patients with ulcerative colitis.4,70 Vitaxin, a humanized antibody to the αVβ3 integrin, inhibited synovial neovascularization in animal models of arthritis, yet little efficacy was observed in a phase II human RA trial.76 Various anti-CD44 antibodies have been tried in arthritis studies.157,158 These and other anti-CAM strategies may be used in other inflammatory conditions including RA.2–465
Chemokine and Chemokine Receptor Targeting
Among traditional DMARDs, sulfasalazine and sulfapyridine inhibited chemokine production by cultured RA synovial explants.159,160 MTX also suppressed chemokine production in the rat adjuvant-induced arthritis (AIA) model.161 In MTX-treated RA patients, high levels of CCL5 correlated with sustained radiologic progression.162 MTX also decreased CCR2 expression on monocytes isolated from RA patients.163 The combination of MTX and leflunomide inhibited the production of CCL2 (MCP-1), CCL17 (TARC), and CCL22 (MDC) in RA.164 Leflunomide itself suppressed CCL2 (MCP-1) and CCL5 (RANTES) levels in RA patients.165 Regarding biologics, most anti-TNF agents exert inhibitory effects on chemokine production. For example, infliximab reduced synovial expression of CXCL8 (IL-8) and CCL2 (MCP-1) in RA patients, which was associated with diminished inflammatory cell ingress into the synovium.166 Treatment of RA patients with infliximab or etanercept resulted in the sustained retention of CXCR3+ T cells in the circulation indicating a clearance of these cells from the synovium.167 In recent studies, infliximab or etanercept also suppressed the release of CXCL1 (GROα), CXCL8 (IL-8), CXCL10 (IP-10), CXCL16, CCL2 (MCP-1), CCL5 (RANTES), CCL20 (MIP-3α), and CX3CL1 (fractalkine).166,168–175 Infliximab also reduced chemokine production in response to Mycobacteria in RA patients, which may have relevance for increased incidence of tuberculosis during anti-TNF therapy.176 Among newer biologics, tocilizumab also acts by suppressing CAM and chemokine production.149 Depletion of B cells by rituximab also interferes with the CXCL8 (IL-8) network.177 Interestingly, antioxidants such as N-acetyl-L cysteine and 2-oxothiazolidine-4-carboxylate inhibited mRNA expression of CXCL8 (IL-8) and CCL2 (MCP-1) by cytokine-pretreated human synovial fibroblasts.178
Regarding direct chemokine receptor inhibition, most human trials using small molecule inhibitors of chemokine receptors failed.179–181 For example, MLN3897, an oral CCR1 antagonist, in combination with MTX had no significant clinical efficacy in RA.179 Similarly, SCH351125180 and AZD5672,181 oral CCR5 inhibitors, did not give any clinical benefit in RA. Yet numerous CCR1, CCR2, and CCR5 antagonists are in clinical development in arthritis, as well as other inflammatory diseases.182–184
A limited number of human antichemokine studies have been done. There has been one trial using an anti-CXCL8 (IL-8) antibody in RA, but results of this trial were not published and the further development of this compound was terminated.9
Antibodies or peptide inhibitors against CXCL4 (PF4), CXCL5 (ENA-78), CXCL8 (IL-8), CXCL10 (IP-10), CXCL16, CCL2 (MCP-1), and CX3CL1 (fractalkine) have been tried successfully in animal models of arthritis,56,67,134,185–187 but none of these agents has yet reached human development. Our group assessed a neutralizing polyclonal anti-CXCL5 antibody administered intravenously to rats using the AIA model. The antibody injected before the onset of arthritis attenuated the severity of the disease. This antibody also prevented the ingress of IL-1-expressing leukocytes into the synovium.186 Regarding chemokine receptor targeting in rodents, CXCR2, CXCR4, CCR1, and CCR5 antagonists have been tried in these animal models.9,134,188,189 Bicyclam, also known as AMD3100, a highly selective antagonist of CXCL12 (SDF-1)/CXCR4, inhibited inflammation and angiogenesis.189 The failure of numerous oral CCR1 and CCR5 antagonists in human trials is discussed earlier.
Some studies have addressed the use of combined chemokine blockade. For example, a combination of CCL2 (MCP-1) and CXCL1 (GROα) inhibition resulted in more pronounced arthritis suppression than CCL2 blockade alone in a murine AIA model.190 Certainly, there may be increased toxicity using combined strategies.9 Hence chemokine or chemokine receptor blockade using antibodies or other inhibitors may be promising for future therapies.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


